


Última actualización:
September 27, 2021
Años publicados: 1986, 1987, 1990, 1991, 1992, 1996, 1997, 1998, 1999, 2000, 2002, 2007, 2017
NORD agradece a Xenia Chepa-Lotrea, NIH/Instituto Nacional de Investigación del Genoma Humano, candidata a doctora en medicina (MD) en la Facultad de Medicina de la Universidad de Georgetown, y a Clair Francomano, MD, Directora de Genética de Adultos y Directora del Centro de Investigación y Atención Clínica de la Fundación Nacional Ehlers-Danlos, Greater Baltimore Medical Center, Harvey Institute for Human Genetics, por su asistencia en la preparación de este informe en inglés. El informe en ingles fue traducido al español y modificado por Gioconda Alyea, medica genetista brasileira el 20 de mayo del 2023.
Los síndromes de Ehlers-Danlos (SED) son un grupo de enfermedades causadas por diferentes defectos genéticos en el colágeno. El colágeno es uno de los principales componentes estructurales del cuerpo. El colágeno es una proteína resistente y fibrosa, y sirve como un bloque de construcción esencial tanto para fortalecer el tejido conectivo (por ejemplo, huesos) como para proporcionar flexibilidad donde sea necesario (por ejemplo, cartílago). Los problemas observados en pacientes con SED pueden deberse a la escasa resistencia del colágeno, o a la ausencia de cantidades suficientes de colágeno estructuralmente normal. Las principales complicaciones que se observan en el SED involucran la piel, los músculos, el esqueleto y los vasos sanguíneos.
Los síndromes de Ehlers-Danlos fueron clasificados originalmente bajo once designaciones de números romanos (EDS I -EDS XI), basándose principalmente en los síntomas y el modo de herencia. Más tarde, EDS se clasificó en seis subtipos según los rasgos característicos de cada tipo. En 2017 se publicó la Clasificación Internacional de los Síndromes de Ehlers-Danlos, en la que se reconocen trece subtipos descriptivos. La Clasificación Internacional de 2017 describe más recientemente una clasificación basada en causas genéticas subyacentes (Grupo A-F) que se utiliza con fines de investigación.
Todos los tipos de SED tienen algunas características comunes como una piel elástica, frágil o floja, heridas que se tardan en curar, desarrollo de moretones fácilmente, cicatrices que son anchas y delgadas como papel (cicatrices de papel de cigarrillo), fragilidad delos vasos de la sangre, tendencias a episodios de sangrado, articulaciones que son muy móviles y propensión a tener lesiones en las articulaciones como dislocaciones y subluxaciones que pueden ocurrir de forma espontánea.
Cada subtipo de EDS es causado por un cambio genético (variante patogénica o mutación) distinto. En el tipo hipomóvil todavía no se ha identificado una mutación.
Hay una serie de criterios clínicos para cada subtipo del SED que permiten hacer un diagnóstico. De esta forma, las señales y los síntomas de una persona en la que se sospecha el SED se pueden comparar con los criterios mayores y menores para identificar a qué subtipo se ajustan más. El diagnóstico definitivo de todos los subtipos de SED en los que se ha identificado la mutación de los genes (todos excepto el SED hipermóvil) requiere la realización de pruebas genéticas de confirmación para identificar la variante causante en cada subtipo.
SED Clásico (cSED): cSED (anteriormente EDSI y EDSII) está asociado con los problemas principales descritos anteriormente, hiperextensibilidad de la piel, laxitud de las articulaciones y vasos sanguíneos frágiles. Las señales y síntomas incluyen:
SED similar al tipo clásico (clSED): El curso clínico de este tipo es similar al del tipo clásico pero las causas geneticas son diferentes. Hay dos tipos, tipo 1 y tipo 2.
SED tipo cardiaco-valvular (cvSED): cvSED es un subtipo raro de SED en el que los pacientes pueden tener signos menores de SED con defectos severos en su arteria aorta, que requieren intervenciones quirúrgicas.
SED tipo vascular (vSED): El tipo vascular (anteriormente SED IV) se caracteriza por:
Los embarazos deben considerarse de mayor riesgo y vigilar de cerca las rupturas arteriales y uterinas. Además, las personas afectadas pueden ser propensas a tener ciertas complicaciones durante y después de los procedimientos quirúrgicos, como la separación de las capas de una herida quirúrgica (dehiscencia).
La expectativa de vida promedio para vSED es de 50 años, pero con una vigilancia cuidadosa y el manejo de las complicaciones, esta edad puede prolongarse.
SED Tipo de hipermovilidad (hSED): hSED (anteriormente SED III) generalmente es una forma menos grave de los SED. Por ejemplo, la dilatación de la raíz aórtica suele ser mínima y no hay aumento de tener disecciones. Las principales complicaciones de los pacientes con hSED son problemas de los músculos y de los huesos.
SED tipo artrocalasia (aSED): aSED (anteriormente SED VII, A y B) se asocia con el riesgo de por vida de la dislocación de múltiples articulaciones al mismo tiempo. Las personas con esta condición tienen dificultades para locomoverse. Es importante identificar a las personas afectadas lo antes posible en la vida, ya que conlleva consecuencias de discapacidad física con la edad. Los recién nacidos pueden mostrar un tono muscular muy disminuido (hipotonía muscular severa) y una dislocación bilateral de las caderas al nacer y pueden ser difíciles de distinguir del kSED.
SED tipo dermatosparexia (dSED): Los pacientes con dSED (anteriormente SED VIIC) tienen una característica que ayuda a diferenciar este tipo de SED de otros, que es una piel flácida y redundante, pareciendo más un tipo de cutis laxa que un tipo de SED. La piel también es extremadamente frágil. El rostro parece tener un aspecto característico, la piel de la cara es floja y colgada, la parte blanca de los ojos (esclerótica) puede ser azulada, hay pliegues de piel en los parpados superiores de los ojos (epicantos), las esquinas externas de los ojos son inclinadas hacia abajo, y la mandíbula es pequeña (micrognatia). Otras características incluyen:
SED tipo cifoescoliótico (kSED): Se caracteriza por un tono muscular bajo presente al nacimiento (hipotonía muscular congénita), curvatura anormal de la columna (cifoescoliosis congénita o de inicio temprano) que puede ser progresiva o no, y una excesiva movilidad de las articulaciones (hipermovilidad articular generalizada) con luxaciones/subluxaciones (en particular de hombros, caderas y rodillas). Síntomas característicos también incluyen:
También puede haber:
A pesar de la escoliosis progresiva, la sobrevida es normal, pero los adultos más gravemente afectados pueden perder la capacidad de caminar entre los 20 y los 30 años y es importante vigilar que su escoliosis no cause problemas con la respiración. Las pruebas moleculares son necesarias para la confirmación diagnóstica.
Síndrome de córnea frágil (SCF o SBC, por sus siglas en inglés): SBC es una variante de SED que también involucra los ojos. Las personas afectadas tienen un riesgo de tener ruptura de la córnea después de sufrir lesiones menores con cicatrización, degeneración de la córnea (queratocono) y protrusión de la córnea (queratoglobo). Puede haber esclerótica azul.
SED tipo espondilodisplásico (spSED): Es una variante de SED con problemas óseos que afecta principalmente a la columna vertebral y las manos. La presentación clínica puede incluir retraso en el crecimiento, baja estatura, ojos saltones con esclerótica azulada, piel arrugada de las palmas de las manos, atrofia (desgaste) de los músculos en la base del pulgar (músculos tenares) y dedos afilados.
SED tipo musculocontractural (mcSED): Este subtipo se asocia especialmente con retraso en el desarrollo y debilidad muscular además de hipotonía. Las personas afectadas con este tipo a menudo tienen defectos estructurales faciales y craneales, contracturas congénitas de los dedos, cifoescoliosis severa, hipotonía muscular, deformidad del pie zambo y problemas de los ojos.
SED tipo miopático (mSED): Se caracteriza por hipotonía muscular evidente al nacer y por enfermedad de los músculos (miopatía). También puede haber escoliosis y sordera de tipo neurosensorial y tiene muchas características parecidas con la forma cifoescoliótica de EDS.
SED tipo periodontal (pSED): El tipo pSED (anteriormente SED VII) tiene hallazgos que incluyen enfermedad de los tejidos que rodean y sostienen los dientes (enfermedad periodontal), lo que puede provocar la pérdida prematura de dientes.
Algunos subtipos de SED incluidos en el sistema original de clasificación de enfermedades ya no forman parte de la clasificación actual (por ejemplo, el tipo anteriormente conocido como «SED tipo IX» se ha redefinido como «síndrome del cuerno occipital» y el tipo «SED XI» ahora se conoce como «síndrome de hiperlaxitud familiar»).
Algunos de los genes asociados con SED proporcionan instrucciones para fabricar (codifican) diferentes subtipos de colágeno (COL1A1, COL1A2, COL1A3, COL5A1 y COL5A2). Otros genes (ADAMTS2, PLOD1 y TNXB) codifican proteínas asociadas con el procesamiento del colágeno o que interactúan con el colágeno. Los defectos en estos genes se han asociado con diferentes subtipos de SED.
SED tipo clásico: Se hereda de forma autosómica dominante y es causado por mutaciones en dos genes: COL5A1 y COL5A2. COL5A1 codifica la proteína ‘pro-alpha1(V)chain’ y COL5A2 codifica la ‘pro-alpha2(V)chain’. La designación «pro-» indica que su producto final debe recibir la acción de una enzima (una proteína especial) para ser activada. El procolágeno es el producto de tres cadenas de proteínas. El procolágeno es procesado por enzimas, situadas fuera de la célula, para que se active. El colágeno final se asociará en fibrillas con colágeno tipo 1 y funcionará para determinar el ancho de las fibrillas de colágeno tipo 1.
SED similar al tipo clásico (clSED): Se hereda de forma autosómica recesiva. Es causado por mutaciones en el gen TNXB que produce una proteína, Tenascin-x, que sirve para mantener la integridad del andamio en el que se deposita el colágeno. Tenascin-x también funciona para regular la estabilidad de las fibras elásticas del cuerpo.
SED tipo valvular cardiaco (cvSED): Se hereda de forma autosómica recesiva y también es asociado con mutaciones en el gen COL1A2. COL1A2 codifica la cadena pro-apha2(I). Dos cadenas pro-alfa1(I) (codificadas por COL1A1) y una cadena pro-apha2(I) (codificada por COL1A2) se asocian para formar fibrillas de procolágeno tipo 1.
SED tipo vascular (vSED): Se hereda de forma autosómica dominante y generalmente es causado por mutaciones en el gen COL3A1. COL3A1 codifica la cadena pro-alfa1(III). Tres de estos productos se asocian para formar procolágeno tipo III. El colágeno tipo III maduro se ensambla en fibrillas largas y delgadas. El entrecruzamiento le da una fuerza importante a este subtipo de colágeno. Algunos pacientes con vSED tienen mutaciones en el gen COL1A1 (descritas anteriormente).
SED tipo de hipermovilidad (hSED): Se hereda de forma autosómica dominante pero no se han identificado los genes que la causan. Hay un pequeño número de personas afectadas con hSED que tienen una deficiencia de tenascina-x, una proteína codificada por el gen TNXB.
SED tipo artrocalasia (aSED): Se hereda de forma autosómica dominante. Este subtipo es causado por mutaciones en el gen COL1A1 o el gen COL1A2. COL1A1 codifica la cadena pro-apha1(I). COL1A2 codifica la cadena pro-apha2(I) (descrita anteriormente).
SED tipo dermatosparaxis (dSED): Se hereda de forma autosómica recesiva y está asociado con mutaciones en el gen ADAMTS2. La enzima codificada por este gen modifica los productos de colágeno. Divide las cadenas cortas de aminoácidos de las moléculas de procolágeno en colágeno maduro.
SED tipo cifoescoliótico (kSED): Se hereda de forma autosómica recesiva y es causado por mutaciones en los genes PLOD1 o FKBP14. Las mutaciones en PLOD1 resultan en una deficiencia de actividad de la enzima procolágeno-lisina, 2-oxogluterato 5-dioxigenasa 1, también conocida como lisil hidroxilasa 1. Esta enzima hidroxilasa convierte el aminoácido lisina en hidrolisina. La hidrolisina es un aminoácido modificado esencial para formar enlaces cruzados entre cadenas individuales de colágeno. El gen FKBP14 codifica la proteína 14 de unión a FK506, que no tiene una función claramente definida en la célula.
Síndrome de córnea frágil (SCF): Hay dos tipos de SCF, ambos heredados de forma autosómica recesiva. El SCF tipo 1 es causado por mutaciones en el gen ZNF469. Se cree que la proteína de dedo de zinc 469 actúa para la síntesis u organización de la fibra de colágeno. El tipo 2 es causado por mutaciones en el gen PRDM5. La proteína codificada, PR/SET dominio 5, regula la síntesis de proteínas.
SED tipo espondilodisplásico (spSED): Se hereda de forma autosómica recesiva y es causado por mutaciones en el gen SLC39A13, que codifica el miembro 13 de la familia 39 de portadores de solutos de productos proteicos, que es responsable del transporte de zinc a la célula. El zinc es un elemento metálico y esencial para el funcionamiento saludable de los tejidos conectivos. Este subtipo también puede ser causado por mutaciones en los genes B4GALT6 y B4GALT7. La familia de genes beta-1,4-galactosiltransferasa está asociada con la síntesis de diferentes estructuras sacáridas y glicosiladas.
SED tipo musculocontractural (mcSED): Se hereda de forma autosómica recesiva y es causado por mutaciones en dos genes, el gen CHST14 y el gen DSE. El gen CHST14 codifica la enzima carbohidrato sulfotransferasa 14, que está involucrada en varias reacciones químicas que participan en la transferencia de grupos sulfato entre diferentes moléculas. El gen DSE codifica la enzima dermatán sulfato epimerasa, que es importante en la producción de dermatán sulfato (también conocido como condroitín sulfato B), que sirve para llenar los huecos del tejido conectivo, dando cohesión y estabilidad.
SED tipo miopático (mSED): Se hereda de forma autosómica dominante o autosómica recesiva. Es causado por mutaciones en el gen COL12A1, que codifica el colágeno tipo XII, que se asocia con el colágeno tipo I. También puede ser causado por mutaciones en el gen FKBP14 que codifica una proteína que todavía no tiene una función claramente definida en la célula.
SED tipo periodontal (pSED): Se hereda de forma autosómica dominante y es causado por mutaciones en los genes C1R y C1S, que codifican subcomponentes del complemento, una sustancia importante para la función inmune.
Herencia
Como comentado antes, los SED se puede heredar como una condición genética dominante o recesiva. Los genes se reciben en conjuntos de dos, uno del padre y otro de la madre.
Los trastornos genéticos dominantes ocurren cuando sólo se necesita una sola copia (alelo) de un gen alterado (mutado) para causar una enfermedad en particular. El gen anormal (mutado) se puede heredar de cualquiera de los padres. El riesgo de transmitir el gen mutado de un padre afectado a su descendencia es del 50% por cada embarazo. El riesgo es el mismo para hombres y mujeres.
En algunas personas, el trastorno se debe a una mutación genética espontánea (de novo) que se produce en el óvulo o el espermatozoide. En tales situaciones, el trastorno no se hereda de los padres.
Los trastornos genéticos recesivos ocurren cuando un individuo hereda dos copias de un gen mutado para el mismo rasgo, una de cada padre. Si un individuo hereda un gen normal y un gen de la enfermedad, la persona será portadora de la enfermedad, pero por lo general no mostrará síntomas. El riesgo de que dos padres portadores transmitan el gen alterado y tengan un(a) hijo(a) afectado(a) es del 25 % en cada embarazo. El riesgo de tener un(a) hijo(a) portador(a) como los padres es del 50% con cada embarazo. La posibilidad de que solamente se transmitan los genes normales de ambos padres es del 25%. El riesgo es el mismo para hombres y mujeres.
Cuando a alguien en la familia se le diagnostica con SED, es importante contactar a un médico para una evaluación adicional y para determinar el modo de herencia en la familia.
Las estimaciones informadas para la incidencia de todos los tipos de SED oscilan entre 1/2500 y 1/5000 nacimientos. Se estima que hSED afecta a 1/10.000-1/15.000. Se estima que cSED afecta a 1/20.000-1/40.0000.
Debido a que existen personas afectadas con SED que tienen manifestaciones articulares y cutáneas leves estas personas pueden no buscar atención médica y no son diagnosticadas por lo que es difícil determinar la verdadera frecuencia de las mutaciones de SED en la población general. hSED, clSED y vSED son los subtipos más comunes. Otros subtipos (kSED aSED y dSED) son mucho menos comunes.
El SED generalmente se diagnostica en función del historial del paciente y de los señales y los síntomas presentes. Las pruebas genéticas pueden facilitar el diagnóstico de algunos subtipos. El análisis microscópico electrónico de muestras de tejido también puede mostrar, a veces, las anomalías características en la estructura del colágeno que se observan en el SED.
Las señales y los síntomas del SED pueden manifestarse durante la niñez. Sin embargo, dependiendo de la forma y la gravedad, la edad del diagnóstico varía ampliamente.
La evaluación clínica de personas con sospecha de SED o con SED ya diagnosticado generalmente incluye evaluaciones para detectar y determinar el grado de hiperextensibilidad de la piel y las articulaciones. Por ejemplo, los médicos pueden medir la hiperextensibilidad de la piel estirando con cuidado la piel en un sitio neutral hasta el punto de resistencia, y la hiperextensibilidad articular puede evaluarse utilizando una escala de calificación clínica (es decir, la escala de Beighton).
A menudo, se utilizan pruebas de imagen especializadas, como la tomografía computarizada (TC), la resonancia magnética nuclear (RMN) y la ecocardiografía, para detectar y caracterizar el prolapso de la válvula mitral y la dilatación aórtica. Durante una tomografía computarizada, una computadora y rayos X crean una película que muestra imágenes transversales de ciertas estructuras corporales. La resonancia magnética utiliza un campo magnético para crear imágenes transversales de órganos y tejidos. Durante un ecocardiograma, las ondas de sonido se dirigen hacia el corazón, lo que permite a los médicos estudiar la función y el movimiento cardíacos.
Además, en algunas personas con SED, se pueden usar estudios de rayos X especializados para caracterizar bultos móviles redondos (esferoides calcificados) debajo de la piel, para detectar y determinar la extensión de la curvatura espinal anormal (escoliosis y/o cifosis) y/o reducción de la masa ósea (ostepenia) (por ejemplo, en aquellos con SED tipos de artrocalasia o cifoescoliosis), y/o para confirmar y caracterizar ciertas otras anomalías.
El análisis genético es útil en el diagnóstico de muchos subtipos de SED, ya sea proporcionando un hallazgo positivo (como por ejemplo, identificando mutaciones en COL5A1 para los pacientes con cSED) o un hallazgo negativo. (cuando no se identifican mutaciones). En el tipo hSED aún no se ha identificado una causa genética por lo que es importante descartar mutaciones que causen otros tipos de SED.
Un diagnóstico de kSED también se puede confirmar mediante una prueba de laboratorio en una muestra de orina y la proporción extrapolada de enlaces cruzados de desoxipiridinolina a piridinolina, o en una muestra de biopsia de piel y la medición de la actividad de la enzima lisil hidroxilasa de las células de fibroblastos de la piel.
El cuidado de los pacientes con SED generalmente se hace tomando medidas preventivas contra complicaciones graves o potencialmente mortales. Las principales complicaciones que se observan en el SED involucran la piel, los sistemas musculoesquelético y cardiovascular.
La piel del paciente es aterciopelada, fina, suelta y elástica. Esto predispone a los pacientes a dificultades con la cicatrización de heridas. Tanto en las heridas accidentales como durante los procedimientos quirúrgicos, se deben aplicar puntos profundos. Se colocan puntos superficiales para realinear cuidadosamente la piel y para evitar cicatrices. Los puntos de sutura también se dejan durante largos períodos de tiempo para que el tejido cicatricial en formación tenga tiempo de fortalecerse. Se puede recomendar ácido ascórbico (vitamina C) para ayudar a reducir los moretones fáciles que se ven en los SED.
Las articulaciones hipermóviles se dislocan fácilmente. Con cada luxación, las luxaciones subsiguientes son cada vez más probables, por lo que la prevención es particularmente importante para la calidad de vida. Deben evitarse los deportes pesados, el levantamiento de pesas y otros esfuerzos extenuantes debido al riesgo de trauma.
La fragilidad de los vasos sanguíneos aumenta el riesgo de hemorragias y de disecciones graves. La presión arterial alta (hipertensión) ejerce una presión adicional sobre los vasos de la sangre que ya están frágiles y esto aumenta el riesgo de complicaciones. Por eso, se deben realizar exámenes regulares para verificar la presión y detectar la enfermedad arterial y se debe iniciar el tratamiento lo antes posible.
Los mejores enfoques para la detección son mediante tecnología no invasiva: ultrasonido, resonancia magnética o tomografía computarizada. La arteriografía, la colonoscopia y otros procedimientos de detección invasivos similares deben considerarse cuidadosamente para evaluar el beneficio frente al riesgo.
También se debe considerar cuidadosamente la cirugía para afecciones que no ponen en peligro la vida. Los embarazos deben ser controlados por obstetras que estén bien capacitados para tratar embarazos de alto riesgo. El parto puede progresar muy rápidamente en pacientes con SED y aún no está claro si existe una ventaja para las madres al dar a luz por vía vaginal o por cesárea. Las futuras madres con problemas cardiacos o dilataciones conocidas de la raíz aórtica deben hacerse ecocardiogramas cada trimestre para observar posibles problemas.
Todas las personas afectadas por EDS deben buscar atención médica inmediata por cualquier dolor repentino o inexplicable y considerar usar un brazalete MedicAlert para comunicar su estado como paciente si pierden el conocimiento.
Los pacientes con hSED pueden beneficiarse especialmente de la fisioterapia, el ejercicio de baja resistencia y los dispositivos de asistencia como aparatos ortopédicos, sillas de ruedas y scooters. Los utensilios de escritura cómodos y un colchón de bajo estrés son importantes para mejorar el dolor. El manejo del dolor se adapta al individuo.
Para mejorar la densidad ósea se hace la fisioterapia y el ejercicio de baja resistencia, y se puede suministrar el calcio y la vitamina D. Las investigaciones de densidad ósea DEXA deben realizarse a cada dos años.
Las complicaciones gastrointestinales y psicológicas también se manejan según las necesidades de cada individuo.
Los pacientes con kSED deben someterse a exámenes oculares de rutina ya que corren el riesgo de ruptura del globo ocular, desprendimiento de retina y glaucoma. Los pacientes con dSED pueden beneficiarse de vendajes protectores sobre las áreas expuestas, como la piel de los codos y las rodillas.
El sitio en la red de Clinical Trials, desarrollado por los Institutos Nacionales de la Salud, proporciona información sobre las investigaciones clínicas.
Usted puede ver las investigaciones sobre los síndromes de Ehlers-Danlos en el siguiente enlace: Clinicaltrials.gov Use el término “Ehlers-Danlos syndrome” para ver los estudios disponibles. Recomendamos que comparta esta información con los médicos para que analicen los estudios y determinen la indicación de la participación en algún estudio. (en inglés)
Para obtener información sobre los ensayos clínicos en Europa, póngase en contacto con: Clinicaltrialsregister.eu
Jones KL, ed. Smith’s Recognizable Patterns of Human Malformation. 5th ed. Philadelphia, PA: W. B. Saunders Co: 1997:482-83.
Bennett JC, Plum F, eds. Cecil Textbook of Medicine. 20th ed. Philadelphia, PA: W.B. Saunders Co; 1996:1120-22.
Buyse ML, ed. Birth Defects Encyclopedia. Dover, MA: Blackwell Scientific Publications; For: The Center for Birth Defects Information Services Inc; 1990:198, 610-11, 1269-70.
Malfait F, et al. The 2017 International Classification of the Ehlers-Danlos Syndromes. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):8-26. https://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31552/full
Henderson Sr FC,et al. Neurological and spinal manifestations of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):195-211. https://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31552/full
Bowen JM, et al. Ehlers-Danlos syndrome, classical type. Am J Med Genet C Semin Med Genet. 2017 Feb 13, Epub ahead of print. https://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31548/full
Chopra P, et al. Pain management in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017 Feb 13, Epub ahead of print. https://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31554/full
Ericson WB Jr and Wolman R. Orthopaedic management of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017 Feb 13, Epub ahead of print. https://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31551/full
Hakin A, et al. Cardiovascular autonomic dysfunction in Ehlers-Danlos syndrome-hypermobile type. Am J Med Genet C Semin Med Genet. 2017 Feb 13, Epub ahead of print. https://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31543/full
Bulbena A, et al. Psychiatric and psychological aspects in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017 Feb 10, Epub ahead of print. https://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31544/full
Fikree A, et al. Gastrointestinal involvement in the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017 Feb 10, Epub ahead of print. https://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.31546/full
Burkitt Wright EM, Porter LF, Spencer HL, et al. Brittle cornea syndrome: recognition, molecular diagnosis and management. Orphanet Journal of Rare Diseases. 2013;8:68. doi:10.1186/1750-1172-8-68.
Giunta, C, et al. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome–an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am. J. Hum. Genet. 2008;82:290-1305.
McDonnell et al. Echocardiographic findings in classical and hypermobile Ehlers-Danlos syndromes. Am J Med Genet A. 2006 Jan 15:140(2):129-36.
Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med. 2001;345:1167-75.
Pepin M, et al. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. New Engl J Med. 2000;342:673-80.
Pyeritz RE, Ehlers-Danlos syndrome. New Engl J Med. 2000:342:730-32.
Beighton P, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31-7.
Michalickova K, et al. Mutations of the alpha2(V) chain type V collagen impair matrix assembly and produce Ehlers-Danlos syndrome type I. Hum Mol Genet. 1998;7:249-55.
Richards AJ, et al. A single base mutation in COL5A2 causes Ehlers-Danlos syndrome type II. J Med Genet. 1998;35:846-48.
Byers PH, et al. Ehlers-Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am J Med Genet. 1997;72:94-105.
De Paepe A, et al. Mutations in the COL5A1 gene are causal in the Ehlers-Danlos syndromes I and II. Am J Hum Genet. 1997;60:547
Burrows NP, et al. The gene encoding collage alpha1(V)(COL5A1) is linked to mixed Ehlers-Danlos syndrome type I/II. J Invest Dermatol. 1996;106:1273-76.
Loughlin J, et al. Linkage of the gene that encodes the alpha 1 chain of type V collagen (COL5A1) to type II Ehlers-Danlos syndrome (EDS II). Hum Mol Genet. 1995;4:1649-51.
North KN, et al. Cerebrovascular complications in Ehlers-Danlos syndrome type IV. Ann Neurol. 1995;38:960-64.
Narcisi P, et al. A family with Ehlers-Danlos syndrome type III/articular hypermobility syndrome has a glycine 637 to serine substitution in type III collagen. Hum Mol Genet. 1994;3:1617-20.
Superti-Furga A, et al. Microangiopathy in Ehlers-Danlos syndrome type IV. Int J Microcirc Clin Exp. 1992;11:241-47.
Wenstrup RJ, et al. Ehlers-Danlos type VI: clinical manifestations of collagen lysyl hydroxylase deficiency. J Pediat. 1989;115:405-09.
Superti-Furga A, et al. Ehlers-Danlos syndrome type IV: a multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability, and processing of type III procollagen. J Biol Chem. 1988;263:6226-32.
Reardon W, et al. The natural history of human dermatosparaxis (Ehlers-Danlos syndrome type VIIC). Clin Dysmorphol. 1995;4:1-11Beighton P, et al. International nosology of heritable disorders of connective tissue, Berlin, 1986. Am J Med Genet. 1988;29:581-94.
Minor RR, et al. Defects in the processing of procellagen to collagen are demonstrable in cultured fibroblasts from patients with the Ehlers-Danlos and osteogenesis imperfecta syndromes. J Biol Chem. 1986;261:10006-14.
Tsipouras P, et al. Ehlers-Danlos syndrome type IV: cosegregation of the phenotype to a COL3A1 allele of type III procollagen. Hum Genet. 1986;74:41-46.
Beighton P, et al. X-linked Ehlers-Danlos syndrome type V: the next generation. Clin Genet. 1985;27:472-78.
Francoman C and Bloom L. The 2017 EDS Classification, Your Questions Answered. [Video webinar]. https://ehlers-danlos.com/wp-content/uploads/QandA-2.pdf. Accessed September 27, 2017.
Malfait F, Wenstrup R, De Paepe A. Ehlers-Danlos Syndrome, Classic Type. 2007 May 29 [Updated 2018]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1244/
Levy HP. Ehlers-Danlos Syndrome, Hypermobility Type. 2004 Oct 22 [Updated 2016 Mar 31]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1279/ Accessed September 27, 2017.
Pepin MG, Murray ML, Byers PH. Vascular Ehlers-Danlos Syndrome. 1999 Sep 2 [Updated 2018 June 21]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1494/
Yeowell HN, Steinmann B. Ehlers-Danlos Syndrome, Kyphoscoliotic Form. 2000 Feb 2 [Updated 2018 October 18]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1462/
Usted puede aprender más sobre el síndrome de Ehlers-Danlos en los siguientes sitios en la red:
Las siguientes fuentes de información en inglés también pueden ser de utilidad:
Visite también nuestra página de NORD en inglés: Ehlers-Danlos syndromes.
Cuando se tiene una enfermedad rara o poco frecuente es muy importante encontrar a un médico que tenga experiencia en el diagnóstico y en el manejo. De forma general, se recomienda que las personas con enfermedades raras busquen ser atendidas en centros médicos universitarios o terciarios ya que es más probable que los médicos que trabajan en estos centros hayan visto casos similares o tengan interés en la investigación, además de que cuentan con equipos de múltiples especialistas que trabajan en conjunto.
NORD tiene una lista de centros de excelencia en enfermedades raras que incluye muchos de los mejores centros médicos y académicos de los Estados Unidos. Recomendamos que los pacientes compartan esta información con sus médicos para que sean referidos al centro más adecuado y conveniente. Esta lista está en expansión.
Para aprender más sobre NORD visite el siguiente enlace: NORD en Español.
Nota: El sitio web de la Organización Nacional de Enfermedades Raras (NORD), sus bases de datos y su contenido tienen derechos de autor de NORD. Ninguna parte del sitio web de NORD, las bases de datos o los contenidos pueden copiarse de ninguna manera, incluidos, entre otros, los siguientes: descarga electrónica, almacenamiento en un sistema de recuperación o redistribución con fines comerciales sin el permiso expreso por escrito de NORD. Sin embargo, por la presente se otorga permiso para imprimir una copia impresa de la información sobre una enfermedad individual para su uso personal, siempre que dicho contenido no se modifique de ninguna manera y el crédito por la fuente (NORD) y el aviso de derechos de autor de NORD estén incluidos en la copia impresa. Cualquier otra reproducción electrónica u otras versiones impresas está estrictamente prohibida.
NORD y la Fundación MedicAlert se han asociado en un nuevo programa para brindar protección a pacientes con enfermedades raras en situaciones de emergencia.
Aprende más https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Asegurarse de que los pacientes y los cuidadores estén equipados con las herramientas que necesitan para vivir su mejor vida mientras manejan su condición rara es una parte vital de la misión de NORD.
Aprende más https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/Este programa de asistencia, primero en su tipo, está diseñado para los cuidadores de un niño o adulto diagnosticado con un trastorno raro.
Aprende más https://rarediseases.org/patient-assistance-programs/caregiver-respite/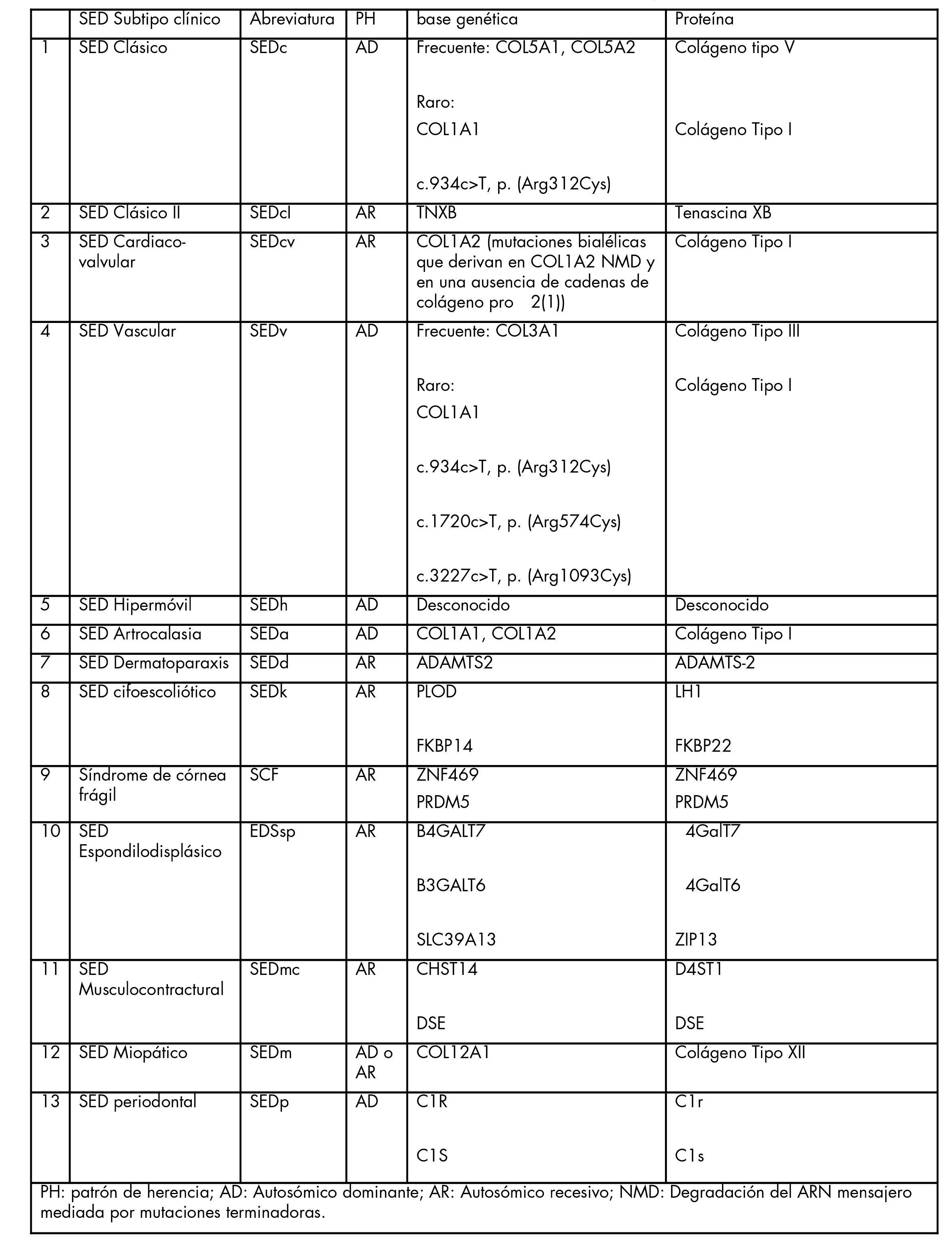{kind=link}