

Última actualización:
7/23/2024
Años publicados: 1984, 1985, 1987, 1990, 1992, 1997, 1999, 2007, 2008, 2011, 2017, 2024
NORD agradece a Anjali Chauhan, directora del programa de asuntos médicos y a Gioconda Alyea, MD (FMG), MS, Organización Nacional de Enfermedades Raras, por su ayuda en la preparación de este informe.
El síndrome de Cri du chat (CdCS o 5p-) es un trastorno genético poco común causado por una pérdida (deleción) en el brazo corto (p) del cromosoma 5.
Los síntomas pueden ser diferentes de persona a persona dependiendo del tamaño exacto y de la ubicación del material genético eliminado. Los síntomas comunes incluyen un llanto que suena como el “maullido de un gato”, rasgos faciales característicos, crecimiento lento y una cabeza más pequeña de lo esperado (microcefalia). Los niños afectados también tienen discapacidad intelectual de moderada a grave, así como retrasos en la adquisición de habilidades (retrasos en el desarrollo) y síntomas adicionales.
En la mayoría de los pacientes, la CdCS es causada por un error genético aleatorio (de novo) que ocurre muy temprano cuando el bebé se desarrolla dentro del útero.
El tratamiento se basa en mejorar los síntomas y puede incluir servicios, terapias y cirugías de intervención temprana.
El síndrome de Cri du chat fue descrito por primera vez en la literatura médica en 1963 por el Dr. Lejeune, quien nombró al trastorno por el distintivo llanto felino. En francés, cri du chat se traduce como «grito del gato».
El síntoma más reconocible es un llanto agudo y estridente característico que está presente durante las primeras semanas de vida. El llanto, que suena como el maullido de un gato, se vuelve menos perceptible a medida que los bebés afectados crecen.
Las señales y los síntomas pueden ser diferentes según el tamaño y la ubicación de la deleción del cromosoma 5. Se han descrito las siguientes señales y síntomas en personas afectadas con el síndrome de cri du chat:
También puede haber:
A medida que los niños afectados crecen, se puede observar un cambio de las señales y de los síntomas:
Además, los niños pueden desarrollar algunos problemas de desarrollo y de conducta como:
El síndrome de cri du chat es un trastorno cromosómico causado por una pérdida parcial (deleción) de parte del brazo corto (p) del cromosoma 5. Los cromosomas son las estructuras que contienen la información genética, o genes, de todos los seres vivos. Los pares de cromosomas humanos están numerados del 1 al 23 y cada cromosoma se divide en dos secciones (brazos) según la ubicación de un estrechamiento (constricción) llamado centrómero. Por convención, el brazo más corto se conoce como “p” y el brazo mayor se conoce como “q”.
Cri du chat también se llama 5p- debido a que falta la información genética contenida en el brazo p del cromosoma 5 (deleción 5p-).
Los síntomas específicos y la gravedad de los síntomas dependen del tamaño y la ubicación de la parte deletada del cromosoma 5p. Los investigadores han determinado que ciertos síntomas pueden estar asociados con regiones específicas en el brazo corto del cromosoma 5. En esas regiones, han identificado varios genes que se cree que desempeñan un papel en el desarrollo del síndrome de cri du chat. Por ejemplo:
Si los investigadores pueden relacionar conjuntos específicos de síntomas y hallazgos (fenotipos) con deleciones específicas en el cromosoma 5p, esto podría ayudar en el diagnóstico y tratamiento de la afección.
En aproximadamente el 10%-15% de los pacientes, el síndrome de cri du chat es causado por una translocación equilibrada que involucra el cromosoma 5p y otro cromosoma. En una translocación equilibrada, piezas de material genético en dos cromosomas diferentes intercambian lugares entre sí (se translocan) por razones desconocidas, pero no se pierde ni se gana ningún material (se equilibra). Por lo tanto, una persona que tiene una translocación equilibrada no tiene ningún problema porque no tiene ninguna ganancia o pérdida de material genético.
Vea una figura del par de cromosomas 5 normal, con sus brazos corto (p) y largo (q) y el par de cromosomas con la deleción del brazo corto (p) que caracteriza el síndrome cri du chat (Fuente: 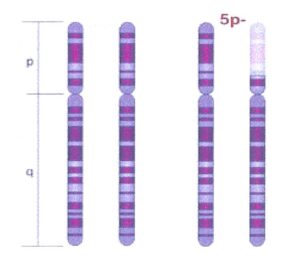
Herencia
La mayoría de los casos de síndrome de cri du chat parecen ocurrir al azar (de novo) por razones desconocidas en una etapa muy temprana del desarrollo embrionario y no se heredan de los padres. La mayoría de las deleciones (80%-90%) son de origen paterno, lo que significa que probablemente ocurren durante la formación de espermatozoides en el padre del niño. Los padres de un niño con una deleción de novo suelen tener cromosomas normales y una probabilidad relativamente baja de tener otro hijo con esta afección.
Sin embargo, si un niño hereda un cromosoma 5 que estuvo envuelto en una translocación equilibrada, ahora tiene un cromosoma 5 al que le falta material genético, lo que puede llevar a desarrollar el síndrome de cri du chat. Además, debido a que un padre puede transmitir ese cromosoma 5, existe una mayor probabilidad de tener otro hijo con esta afección. El análisis cromosómico puede determinar si uno de los padres tiene una translocación equilibrada.
La frecuencia del síndrome de cri du chat oscila entre 1/15.000 y 1/50.000 nacidos vivos. Este síndrome afecta a las mujeres con más frecuencia que a los hombres.
Algunas personas con síndrome de cri du chat pueden no ser diagnosticadas, lo que dificulta determinar la verdadera frecuencia de este trastorno en la población general.
El síndrome de Cri du chat se puede diagnosticar antes o después del nacimiento.
Antes del nacimiento (prenatalmente), la afección se puede diagnosticar mediante un procedimiento llamado amniocentesis. Durante la amniocentesis, se toma una muestra del líquido que rodea al feto en el útero (líquido amniótico) para analizarla. El líquido contiene células del feto, que contienen los cromosomas del feto y se pueden analizar para detectar si hay la deleción en el cromosoma 5.
La amniocentesis se puede combinar con una ecografía. Una ecografía puede revelar características físicas del feto que hacen sospechar una condición genética como el síndrome de cri du chat.
En los recién nacidos, el diagnóstico de síndrome de cri du chat puede sospecharse basándose en una evaluación clínica detallada que identifique las señales y los síntomas característicos (p. ej., llanto de gato).
El diagnóstico se confirma con estudios cromosómicos (cariotipo) que revelan una deleción en el brazo corto del cromosoma 5.
Otras pruebas que pueden usarse para confirmar el diagnóstico de síndrome de cri du chat son la hibridación fluorescente in situ (FISH), la hibridación genómica comparativa ( CGH), o una reacción en cadena de la polimerasa cuantitativa (PCR).
También se pueden realizar estudios cromosómicos a los padres para determinar si uno de ellos tiene una translocación equilibrada.
Se pueden utilizar pruebas adicionales para saber que otros problemas tienen las personas afectadas, como radiografías, para buscar anomalías esqueléticas como la escoliosis.
No hay cura todavía. El tratamiento del síndrome de Cri du chat sedirige a mejorar los síntomas específicos que la persona afectada tenga. Las personas afectadas requieren ser atendidas por muchos especialistas que deben trabajar juntos y de forma coordinada en equipo. El equipo de especialistas puede incluir:
Dado que algunos niños con el síndrome pueden tener sordera neurosensorial, es fundamental realizar pruebas auditivas.
La intervención temprana es crucial para ayudar a los niños a desarrollar su máximo potencial y puede incluir:
Es común que los niños comiencen a recibir estas terapias antes de cumplir un año de edad.
En algunos casos, se puede considerar la cirugía para corregir problemas como defectos cardíacos, estrabismo, escoliosis, y malformaciones como paladar hendido o labio hendido.
La supervivencia de los niños con síndrome de Cri du Chat es generalmente buena. La mayoría de las muertes relacionadas con el síndrome ocurren dentro del primer año de vid y pueden ser debidas a defectos cardiacos a, pero muchas personas afectadas han vivido hasta los 50 años o más.
Otros tratamientos son principalmente sintomáticos y de apoyo, adaptados a las necesidades individuales de cada persona afectada.
El asesoramiento genético es recomendado para los afectados y sus familias para comprender mejor la condición y las posibilidades de tener otros hijos con la misma afección.
Las investigaciones sobre el síndrome de cri du chat continúan, con estudios que sugieren que una educación especial temprana, un entorno hogareño estable y el apoyo familiar pueden mejorar significativamente el desarrollo y la capacidad de comunicación de los niños. En un estudio, se encontró que la mitad de los niños mayores de diez años que recibieron educación especial y vivieron en un entorno de apoyo podían comunicarse adecuadamente.
El sitio en la red de Clinical Trials, desarrollado por los Institutos Nacionales de la Salud, proporciona información sobre las investigaciones clínicas. Usted puede ver las investigaciones sobre la enfermedad de Cri du chat en el siguiente enlace: Clinicaltrials.gov. Use el término “Cri du chat” para ver los estudios disponibles. Recomendamos que comparta esta información con los médicos para que analicen los estudios y determinen la indicación de la participación en algún estudio. (en inglés)
Para obtener información sobre los ensayos clínicos en Europa, póngase en contacto con: Clinicaltrialsregister.eu
Mezoff AG. Cri-du-Chat Syndrome. NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia, PA. 2003:175.
Ullah I, Mahajan L, Magnuson D. A newly recognized association of Hirschsprung disease With Cri-du-chat syndrome. Am J Gastroenterol. 2017;112:185-186.
Nguyen JM, Qualmann KJ, Okashah R, Reilly A, Alexeyev MF, Campbell DJ. 5p deletions: Current knowledge and future directions. Am J Med Genet C Semin Med Genet. 2015 Sep;169(3):224-38.
Rodriguez-Caballero A, Torres-Lagares D, Rodriguez-Perez A, Serrera-Figallo MA, Hernandez-Guisado JM, Machuca-Portillo G. Cri du chat syndrome: A critical review. Oral Patol Oral Cir Bucal. 2010;15:e473-8.
Cerruti Mainardi, P. Cri du Chat syndrome. Orphanet J Rare Dis. 2006;1: 33. https://doi.org/10.1186/1750-1172-1-33
Hill C, Moller JH, Finkelstein M, Lohr J, Schimmenti L. Cri du chat syndrome and congenital heart disease: a review of previously reported cases and presentation of an additional 21 cases from the pediatric cardiac care consortium. Pediatrics. 2006;117:924-7.
Laczmanska I, Stembalska A, Gil J, Czemarmazowicz H, Sasiadek M. Cri du chat syndrome determined by the 5p15.3→pter deletion–diagnostic problems. Eur J Med Genet. 2006;49:87-92.
Mainardi PC, Pastore G, Castronovo C, et al., The natural history of cri du chat syndrome. A report from the Italian Register. Eur J Med Genet. 2006;49:363-83.-9.
Kondoh T, ShimoKawa O, Harada N, Doi T, et al., Genotype-phenotype correlation of 5p- syndrome: pitfall of diagnosis. J Hum Genet. 2005;50:26.
Posmyk R, Panasiuk B, Yatsenko SA, Stankiewicz P, Midro AT. A natural history of a child with monosomy 5- syndrome (cat-cry/cri-du-chat syndrome) during the 18 years of follow-up. Genet Couns. 2005;16:17-25.
Zhang X, Snijders A, Segraves R, et al., High-resolution mapping of genotype-phenotype relationships in cri du chat syndrome using array comparative genomic hybridization. Am J Hum Genet. 2005;76:312-6.
Van Buggenhout GJ, Pijkels E, Holvoet M, et al., Cri du chat syndrome: changing phenotype in older patients. Am J Med Genet. 2000;90:203-15.
Ajitkumar A, Jamil RT, Mathai JK. Cri Du Chat Syndrome. [Updated 2022 Oct 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482460/ Accessed July 10, 2024.
Cri-du-chat syndrome: Medlineplus genetics. MedlinePlus. October 25, 2022. https://medlineplus.gov/genetics/condition/cri-du-chat-syndrome/ Accessed October 3, 2023.
McKusick VA., ed. Online Mendelian Inheritance in Man (OMIM). Baltimore. MD: The Johns Hopkins University; Entry No:123450; Last Update: 1/13/2023. Available at: http://omim.org/entry/123450 Accessed July 22, 2024.
Lal MK. Cri-du-Chat Syndrome. Updated: Mar 01, 2024. Medscape. Available at: http://www.emedicine.com/ped/topic504.htm Accessed July 8, 2024.
Usted puede aprender más sobre esta enfermedad en los siguientes sitios en la red:
Las siguientes fuentes de información en inglés también pueden ser de utilidad:
Vea también nuestra página en inglés de NORD: Cri du chat syndrome.
Cuando se tiene una enfermedad rara o poco frecuente es muy importante encontrar a un médico que tenga experiencia en el diagnóstico y en el manejo. De forma general, se recomienda que las personas con enfermedades raras busquen ser atendidas en centros médicos universitarios o terciarios ya que es más probable que los médicos que trabajan en estos centros hayan visto casos similares o tengan interés en la investigación, además de que cuentan con equipos de múltiples especialistas que trabajan en conjunto.
NORD tiene una lista de centros de excelencia en enfermedades raras que incluye muchos de los mejores centros médicos y académicos de los Estados Unidos. Recomendamos que los pacientes compartan esta información con sus médicos para que sean referidos al centro más adecuado y conveniente. Esta lista está en expansión.
Para aprender más sobre NORD visite el siguiente enlace: NORD en Español.
Nota: El sitio web de la Organización Nacional de Enfermedades Raras (NORD), sus bases de datos y su contenido tienen derechos de autor de NORD. Ninguna parte del sitio web de NORD, las bases de datos o los contenidos pueden copiarse de ninguna manera, incluidos, entre otros, los siguientes: descarga electrónica, almacenamiento en un sistema de recuperación o redistribución con fines comerciales sin el permiso expreso por escrito de NORD. Sin embargo, por la presente se otorga permiso para imprimir una copia impresa de la información sobre una enfermedad individual para su uso personal, siempre que dicho contenido no se modifique de ninguna manera y el crédito por la fuente (NORD) y el aviso de derechos de autor de NORD estén incluidos en la copia impresa. Cualquier otra reproducción electrónica u otras versiones impresas está estrictamente prohibida.

NORD y la Fundación MedicAlert se han asociado en un nuevo programa para brindar protección a pacientes con enfermedades raras en situaciones de emergencia.
Aprende más https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Asegurarse de que los pacientes y los cuidadores estén equipados con las herramientas que necesitan para vivir su mejor vida mientras manejan su condición rara es una parte vital de la misión de NORD.
Aprende más https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/Este programa de asistencia, primero en su tipo, está diseñado para los cuidadores de un niño o adulto diagnosticado con un trastorno raro.
Aprende más https://rarediseases.org/patient-assistance-programs/caregiver-respite/