The animated videos in NORD’s Rare Disease Video Library provide brief introductions to rare disease topics for patients, caregivers, students, professionals and the public. NORD collaborates with medical experts, patient organizations, videographers and Osmosis to develop the videos, which are made possible by individual donations, educational grants and corporate sponsorships. NORD is solely responsible for the content.
La deficiencia de esfingomielinasa ácida (ASMD, por sus siglas en inglés) es un trastorno genético progresivo poco frecuente.
La ASMD es causada por cambios (mutaciones o variantes patogénicas) en el gen SMPD1 que resultan en la deficiencia de la enzima esfingomielinasa ácida, que es necesaria para descomponer (metabolizar) una sustancia grasa (lípido) llamada esfingomielina. Como resultado, la esfingomielina y otras sustancias se acumulan en diversos tejidos del cuerpo.
La ASMD es altamente variable en términos de la edad de inicio, los síntomas específicos y la gravedad de la enfermedad, que pueden variar de forma considerable entre las personas, incluso dentro de una misma familia. De esta forma, este trastorno puede considerarse como un espectro de enfermedades.
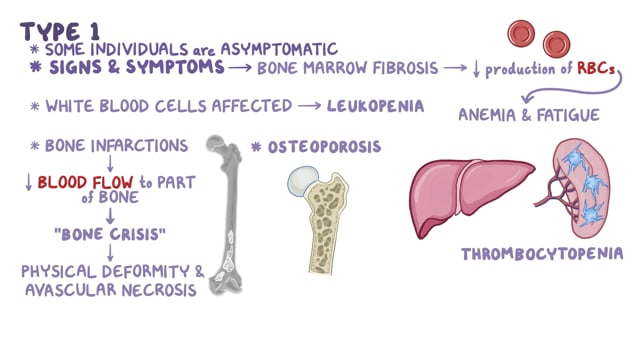
En el extremo más severo del espectro se encuentra un trastorno neurodegenerativo fatal que aparece en la infancia (enfermedad de Niemann-Pick tipo A). En el extremo más leve, las personas afectadas no presentan síntomas neurológicos o estos son mínimos, y la supervivencia hasta la adultez es común (enfermedad de Niemann-Pick tipo B). También existen formas intermedias del trastorno.
La herencia es autosómica recesiva.
Aunque aún no existe cura, en 2022 se aprobó una terapia de reemplazo enzimático, el medicamento Olipudase alfa (Xenpozyme), para tratar los síntomas relacionados con la ASMD que no afectan al sistema nervioso central (SNC).
La deficiencia de esfingomielinasa ácida (ASMD) también se conoce como enfermedad de Niemann-Pick con deficiencia de esfingomielinasa ácida. Existen tres tipos de trastornos conocidos como enfermedad de Niemann-Pick: tipos A, B y C. Estos trastornos se agruparon inicialmente debido a síntomas similares, pero ahora sabemos que son enfermedades diferentes. Los tipos A y B de Niemann-Pick se deben a mutaciones en el gen SMPD1, lo que provoca una deficiencia de la enzima esfingomielinasa ácida (ASM). Por otro lado, la enfermedad de Niemann-Pick tipo C es causada por mutaciones en otros dos genes y no implica una deficiencia enzimática. Por este motivo, el tipo C se considera un trastorno separado y distinto de los tipos A y B.
Tradicionalmente, la ASMD se ha clasificado en dos subgrupos: neuronopática (tipo A) y no neuronopática (tipo B). El término «neuronopática» se refiere a trastornos que dañan las células cerebrales (neuronas). El tipo A suele causar una enfermedad neurodegenerativa grave durante la infancia, mientras que el tipo B generalmente no se considera una enfermedad neurológica. Sin embargo, dado que algunos casos se sitúan entre estos extremos, estas clasificaciones amplias pueden ser engañosas. Algunos investigadores usan el término ASMD tipo B para referirse a todas las formas leves e intermedias, incluidas aquellas con hallazgos neurológicos.