- Donate
- Understanding Rare Disease
- Living with a Rare Disease
- Community Support
- Advancing Research
- Driving Policy
- Get Involved
- Rare Disease News
- Resource Library
- About Us
- For Clinicians & Researchers
- For Patient Organizations


The animated videos in NORD’s Rare Disease Video Library provide brief introductions to rare disease topics for patients, caregivers, students, professionals and the public. NORD collaborates with medical experts, patient organizations, videographers and Osmosis to develop the videos, which are made possible by individual donations, educational grants and corporate sponsorships. NORD is solely responsible for the content.
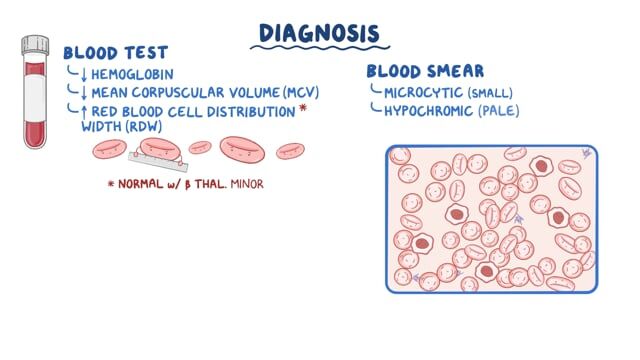
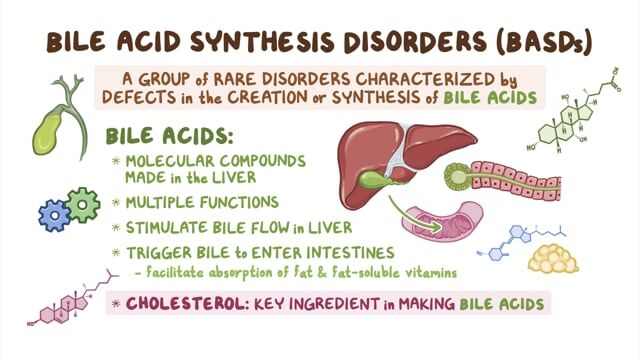
Alagille syndrome (ALGS) is a rare genetic disorder that can affect multiple organ systems of the body including the liver, heart, skeleton, eyes and kidneys. The specific symptoms, organ system involvement and severity of Alagille syndrome can vary greatly from one person to another, even within the same family. Some individuals may have mild forms of the disorder while others may have more serious forms. Common symptoms, which often develop during the first three months of life, include blockage of the flow of bile from the liver (cholestasis), yellowing of the skin and mucous membranes (jaundice), poor weight gain and growth and severe itching (pruritis). Additional symptoms include heart murmurs, congenital heart defects, vertebral (back bone) differences, thickening of the ring that normally lines the cornea in the eye (posterior embryotoxon) and distinctive facial features. Most people with Alagille syndrome have changes (variants or mutations) in one copy of the JAG1 gene. A small percentage (2%) of patients have variants in the NOTCH2 gene. These variants can be inherited in an autosomal dominant pattern, but in about half of cases, the variant occurs as a new change (de novo) in the individual and was not inherited from a parent. The current estimated incidence of ALGS is approximately 1/30,000 –1/45,000.