- Donate
- Understanding Rare Disease
- Living with a Rare Disease
- Community Support
- Advancing Research
- Driving Policy
- Get Involved
- Rare Disease News
- Resource Library
- About Us
- For Clinicians & Researchers
- For Patient Organizations
Last updated: 2/6/2025
Years published: 1986, 1989, 1990, 1993, 1994, 1996, 1997, 2003, 2025
NORD gratefully acknowledges Gioconda Alyea, MD (FMG), MS, National Organization for Rare Disorders for assistance in the preparation of this report.
Summary
Machado-Joseph disease, also known as spinocerebellar ataxia 3 (SCA3) is a rare neurologic condition characterized by progressive problems with movement.1,2
People affected by this condition present with coordination and balance problems (ataxia) with a slowly progressive clumsiness in the arms and legs, a staggering or lurching gait, as well as difficulty with speech and swallowing, vision problems and lower limb spasticity. Additional symptoms may include dystonia (uncontrolled muscle tensing), or symptoms like those of Parkinson’s disease, such as slowness of movement and muscle stiffness (rigidity), chronic pain and vocal cord paralysis. Other people may develop twitching of the face or tongue, neuropathy, sleep disorders, or problems with urination and the autonomic nervous system, which regulates body functions like breathing, digestion, heart rate and blood pressure. A common feature is the impairment of temperature sensation involving the entire body. Other features include cognitive impairments such as problems speaking and remembering and emotional problems such as depression.1,2,3
This condition is caused by changes (variants) in the ATXN3 gene. The ATXN3 gene variants that cause SCA3 involve a DNA segment known as a CAG trinucleotide repeat made up of a series of three amino acids (cytosine, adenine and guanine) that appear multiple times in a row. Normally, the CAG segment is repeated 12 to 43 times within the gene. In people with SCA3, the CAG segment is repeated more than 50 times. Inheritance is autosomal dominant.1,2
There are 3 types of Machado-Joseph disease that are based on the age of onset and severity:1
There is no cure for Machado-Joseph disease but there are treatments to manage symptoms.1
Introduction
Spinocerebellar ataxias (SCA) refer to a group of more than 30 different neurologic conditions characterized by ataxia and other problems and are numbered based on the discovery of the gene variants that cause each type. The term “spinocerebellar ataxia” refers to ataxias that are inherited in an autosomal dominant manner. Machado-Joseph disease (SCA3) is one of these disorders.1
People affected with Machado-Joseph disease may have the following signs and symptoms:1,2,3
As time goes on and the diseases progresses, late-stage symptoms may include:
Symptoms typically begin between around 20 to 50 years of age but can range from childhood to late adulthood. People with earlier-onset MJD are often present with dystonia, while people with later-onset MJD are more likely to show neuropathy and muscle wasting. These symptoms evolve as the condition progresses and individuals often require assistive devices including wheelchairs within 10-15 years of onset.
Based on the age of onset and the severity of the disease, which can vary greatly due to varying CAG repeats, there are three types of Machado-Joseph disease:3
Machado-Joseph Disease (MJD) is caused by variants in the ATXN3 gene. The ATXN3 gene provides instructions for producing an enzyme called “ataxin-3”, which plays a vital role in the ubiquitin-proteasome system. This system degrades damaged or excess proteins in cells. Ataxin-3 is responsible for removing ubiquitin from these proteins before their degradation, allowing ubiquitin to be recycled.1,2,3
The genetic variant causing MJD involves an abnormal CAG trinucleotide repeat expansion within the ATXN3 gene. This segment is made up of a series of three DNA building blocks (cytosine, adenine and guanine – CAG) that appear multiple times in a row. In unaffected people, the CAG sequence repeats between 12 and 43 times, while in affected individuals, the repeat occurs 56 to 86 times. Individuals with a repeat length of 44 to 52, classified as “intermediate,” may or may not develop MJD. The number of repeats correlates directly with the severity of the disease and the age of onset. Fewer repeats are associated with later onset and milder symptoms, typically classified as MJD-I, whereas a higher number of repeats leads to earlier onset and more severe disease as seen in MJD-III. Intermediate cases, known as MJD-II, fall between these extremes.1,2
An increase in the length of the CAG repeat produces an abnormally long version of the ataxin-3 protein, which misfolds and aggregates within the nuclei of cells. These aggregates contain ataxin-3, ubiquitin and unwanted proteins, disrupting normal cellular function. Although these aggregates are observed in healthy cells as well, their accumulation in the neurons of affected individuals is associated with large cell death. Neurons in the brainstem, cerebellum and spinal cord are particularly vulnerable, leading to progressive neuronal degeneration. Over time, this cell loss results in the characteristic symptoms of MJD, including movement and coordination difficulties, as well as other neurological impairments.1,3
Machado-Joseph disease inheritance is autosomal dominant. Dominant genetic disorders occur when only a single copy of a disease-causing gene variant is necessary to cause the disease. The gene variant can be inherited from either parent or can be the result of a new (de novo) changed gene in the affected individual that is not inherited. The risk of passing the gene variant from an affected parent to a child is 50% for each pregnancy. The risk is the same for males and females.2,3
A phenomenon known as anticipation often occurs in MJD. This refers to the observation that the disease manifests earlier and with greater severity in subsequent generations. This is caused by the instability of the CAG repeat expansion during transmission from parent to child. While repeat expansions are more likely with paternal transmission, the paternal bias is not pronounced.3
Machado-Joseph Disease is thought to be the most common type of spinocerebellar ataxia, although all forms of spinocerebellar ataxia are rare. The exact prevalence of SCA3 worldwide is unknown, but it appears to be more common in certain groups, such as people of Portuguese descent, particularly those from the Azores Islands and some indigenous Australian populations.2
In the Azores, SCA3 has a prevalence of approximately 39 per 100,000 people, while in mainland Portugal, it is about 3.1 per 100,000 people. Studies have shown that MJD was first reported in individuals of Azorean ancestry living in the United States.3,4
SCA3 affects slightly more males than females and is one of the most common inherited ataxias in the United States and Canada. It is particularly well-documented in families with a history of Portuguese ancestry with the names Machado, Thomas and Joseph historically linked to its discovery.2,3,4
A diagnosis of spinocerebellar ataxia (SCA) may be suspected based on results from neurological tests and family history. Imaging tests like CT scans or MRIs can help identify changes in the brain, such as shrinkage (atrophy) in the cerebellum or other areas. Other imaging methods can show how the brain is functioning.
To confirm a diagnosis of SCA, genetic testing is needed. This test looks for specific changes (variants) in genes known to cause SCA, such as the ATXN3 gene for Machado-Joseph disease (SCA3). This genetic test is highly accurate.
People who have a parent with Machado-Joseph disease or another form of SCA but don’t have symptoms can choose to have a genetic test to see if they carry the gene variant. This is called presymptomatic testing and can tell if someone is likely to develop the disease later in life.1
Treatment
There is no cure or specific treatment for Machado-Joseph disease. Treatment is based on managing the specific symptoms that the affected person has. It is crucial for people affected by SCA3 to receive care from a multidisciplinary team. This includes neurologists, physiotherapists, speech therapists, nutritionists and mental health professionals. Personalized treatment plans tailored to their specific symptoms and needs can greatly improve quality of life, manage complications and slow the progression of functional decline. Regular evaluations and adjustments to therapies ensure the best possible outcomes, emphasizing the importance of a coordinated, individualized approach to care.3,5
The recommended evaluations after a diagnosis are:3
The treatment of manifestations in SCA3 involves a comprehensive, multidisciplinary approach tailored to individual symptoms:3
Regular assessments are crucial to monitor disease progression.3,5
Innovative methods like siRNAs and antisense oligonucleotides offer hope for targeting ATXN3 gene variants. Small interfering RNA (siRNA) is a synthetic RNA molecule that temporarily silences genes. Studies using lipid nanoparticles, tiny particles composed by lipids that can be used to deliver drugs intravenously to specific sites in the body and synthetic extracellular vesicles have shown efficacy in animal models. These approaches may reduce toxic protein levels, alleviate neuronal inflammation and rescue neuronal function with minimal invasiveness.6,7
Information on current clinical trials is posted on the Internet at https://clinicaltrials.gov/. All studies receiving U.S. Government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Toll-free: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website:
https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, contact:
https://www.centerwatch.com/
For information about clinical trials conducted in Europe, contact:
https://www.clinicaltrialsregister.eu/
NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportOnline Mendelian Inheritance In Man (OMIM) has a summary of published research about this condition and includes references from the medical literature. The summary contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. OMIM is authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine.
View report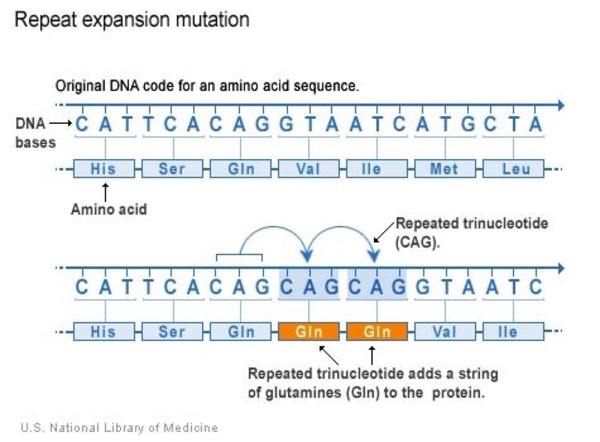{kind=link}