

Última actualización:
April 14, 2016
Años publicados: 1986, 1987, 1990, 1992, 1993, 1997, 1998, 2001, 2011, 2016
NORD agradece sinceramente a Stephen Cederbaum, MD, División de Genética, UCLA, por su asistencia en la preparación de este informe en inglés. El informe en inglés fue traducido al español y modificado por Gioconda Alyea, médica genetista brasileira, el 19 de septiembre del 2024.
La deficiencia de N-acetilglutamato sintetasa (NAGS) es un trastorno genético raro causado por la ausencia completa o parcial de la enzima N-acetilglutamato sintetasa (NAGS). NAGS es una de las seis enzimas que participan en la descomposición y eliminación del nitrógeno del cuerpo, un proceso conocido como el ciclo de la urea.
Las enzimas son proteínas complejas que producen un cambio químico específico. Los trastornos del ciclo de la urea son un grupo de trastornos raros que afectan el ciclo de la urea, una serie de procesos bioquímicos en los que el nitrógeno se convierte en urea y se elimina del cuerpo a través de la orina. El nitrógeno es un producto de desecho del metabolismo de las proteínas. La incapacidad para descomponer el nitrógeno provoca la acumulación anormal de nitrógeno, en forma de amoníaco, en la sangre.
La enzima NAGS es necesaria para activar la enzima carbamoil fosfato sintetasa I, que es la enzima que regula el primer paso del ciclo de la urea, por lo que la deficiencia de NAGS resulta en que no se active el carbamoil fosfato sintetasa I, imposibilitando que se pueda iniciar el ciclo de la urea y resulta en una acumulación excesiva de amoníaco en la sangre (hiperamonemia). El exceso de amoníaco, que es una neurotoxina, viaja al sistema nervioso central a través de la sangre, lo que provoca los síntomas y hallazgos físicos característicos de la deficiencia de NAGS.
Los síntomas incluyen vómitos, rechazo a comer, letargo progresivo y coma. La deficiencia de NAGS se hereda como un rasgo autosómico recesivo.
El tratamiento puede incluir la prevención de la formación excesiva de amoníaco, restricciones dietéticas y medicamentos como Carbaglu® (carbamilglutamato).
Las señales y los síntomas de deficiencia de N-acetilglutamato sintetasa (NAGS) son causados por la acumulación de amoníaco en la sangre debido a la ausencia completa o parcial de la enzima NAGS. La ausencia completa de la enzima NAGS provoca la forma más grave del trastorno, en la que los síntomas aparecen poco después del nacimiento (período neonatal). La falta parcial de la enzima NAGS provoca una forma más leve del trastorno que ocurre más tarde durante la infancia, la niñez o incluso en la edad adulta en algunos casos. Los síntomas específicos pueden variar de una persona a otra.
Forma grave (deficiencia completa de la enzima NAGS):
En los casos más graves, los síntomas aparecen dentro de las 24-72 horas después del nacimiento. Estos incluyen:
Si no se trata, los niveles altos de amoníaco en la sangre pueden llevar a un coma hiperamonémico (un estado profundo de inconsciencia causado por los altos niveles de amonia en la sangre) lo que puede causar:
Es probable que se presenten complicaciones potencialmente mortales si el trastorno no se trata.
Forma más leve (deficiencia parcial de la enzima NAGS):
Algunas personas pueden tener una forma más leve de deficiencia de NAGS, donde los síntomas aparecen más tarde en la infancia, la niñez o incluso en la adultez. Los síntomas pueden incluir:
Incluso con esta forma más leve, existe el riesgo de un coma hiperamonémico y complicaciones potencialmente mortales si la afección no se maneja adecuadamente.
La deficiencia de N-acetilglutamato sintetasa (NAGS) es causada por cambios (variantes patogénicas o mutaciones) en el gen NAGS. Las variantes en este gen resultan en la deficiencia de la enzima N-acetilglutamato sintetasa. Específicamente, la enzima NAGS es un activador de otra enzima del ciclo de la urea, conocida como carbamil fosfato sintetasa (CPS) que es la enzima que se necesita para que se inicie el ciclo de la urea. Los síntomas de la deficiencia de NAGS se desarrollan debido a la falta de esta enzima, que es necesaria para descomponer (metabolizar) el nitrógeno en el cuerpo. La incapacidad para descomponer adecuadamente el nitrógeno resulta en la acumulación anormal de este, en forma de amoníaco, en la sangre (hiperamonemia).
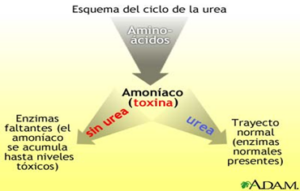
Fuente: https://medlineplus.gov/spanish/ency/esp_imagepages/17167.htm
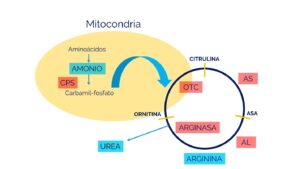
La enzima NAGS produce una molécula llamada N-acetilglutamato, que es esencial para el funcionamiento de la primera enzima del ciclo de la urea, CPS1.
Fuente: https://www.familiasga.com/wp-content/uploads/2019/10/CICLO-DE-LA-UREA.jpg
Herencia
La herencia es autosómica recesiva. Los trastornos genéticos recesivos ocurren cuando un individuo hereda dos copias de un gen anormal para el mismo rasgo, uno de cada padre. Si una persona hereda un gen normal y un gen mutado para la enfermedad, será portadora pero generalmente no mostrará síntomas. El riesgo de que dos padres portadores transmitan el gen alterado (mutado) y tengan un hijo afectado es del 25% en cada embarazo. El riesgo de tener un hijo que sea portador, como los padres, es del 50% en cada embarazo. La probabilidad de que un niño reciba genes normales de ambos padres es del 25%. El riesgo es el mismo para varones y mujeres.
Los padres que son parientes cercanos (consanguíneos) tienen una mayor probabilidad que los padres no relacionados de portar el mismo gen anormal, lo que aumenta el riesgo de tener hijos con un trastorno genético recesivo.
La deficiencia de N-acetilglutamato sintetasa (NAGS) es un trastorno raro que afecta a hombres y mujeres por igual. En la mayoría de los casos, los síntomas comienzan en o poco después del nacimiento. Se estima que la frecuencia de los trastornos del ciclo de la urea es de uno en 30,000 nacimientos. Sin embargo, debido a que los trastornos del ciclo de la urea como la deficiencia de NAGS a menudo no se reconocen, estos trastornos están subdiagnosticados, lo que dificulta determinar la verdadera frecuencia de los trastornos del ciclo de la urea en la población general. La deficiencia de NAGS es el defecto más raro del ciclo de la urea, con una incidencia de menos de uno en 2,000,000 nacimientos.
El diagnóstico de la deficiencia de N-acetilglutamato sintetasa (NAGS), o de cualquier trastorno del ciclo de la urea debe considerarse en cualquier recién nacido con una enfermedad no diagnosticada caracterizada por vómitos, letargo progresivo e irritabilidad. Puede confirmarse con pruebas genéticas que detecten mutaciones en el gen NAGS.
El tratamiento para la deficiencia de N-acetilglutamato sintetasa (NAGS) requiere un equipo de especialistas, que puede incluir pediatras, neurólogos, genetistas, dietistas y médicos especializados en trastornos metabólicos. Para niños con retrasos en el desarrollo, puede ser necesario apoyo adicional de terapeutas ocupacionales, de habla y físicos.
El enfoque principal del tratamiento es:
El tratamiento a largo plazo básicamente es hecho con restricciones dietéticas (limitar proteínas para evitar la acumulación de amoníaco) y con terapia de vías alternativas para eliminar el nitrógeno.
El manejo dietético consiste en limitar el consumo de proteínas mientras se asegura un crecimiento adecuado, especialmente en lactantes. Un plan típico incluye:
Los medicamentos utilizados son fármacos captadores de nitrógeno ayudan a eliminar el exceso de nitrógeno. Estos incluyen:
En 2010, la FDA aprobó Carbaglu® (carbamilglutamato) para reducir los niveles de amoníaco en pacientes con deficiencia de NAGS. Aunque algunas personas afectadas aún necesitan restricciones dietéticas y arginina suplementaria.
Si hay un episodio grave de hiperamonemia, el tratamiento inmediato es fundamental:
En casos pasados, un procedimiento llamado hemodiálisis era necesario si no se respondía al tratamiento o si los pacientes entraban en coma. Sin embargo, la terapia con Carbaglu ha reducido esta necesidad.
Después del diagnóstico, es esencial el cuidado continuo para prevenir la acumulación de amoníaco. Los exámenes de sangre regulares para monitorear los niveles de amoníaco y glutamina pueden ayudar a iniciar el tratamiento antes de que aparezcan los síntomas.
Los estudios recientes en animales de laboratorio indican que la terapia génica podría ser útil para tratar la deficiencia de NAGS. Se han utilizado vectores virales para entregar genes funcionales de NAGS en modelos animales, lo que podría corregir la deficiencia enzimática en el futuro.
La consejería genética puede ser útil para las familias afectadas, proporcionando información sobre el trastorno, patrones de herencia y opciones para la planificación familiar.
El sitio en la red de Clinical Trials, desarrollado por los Institutos Nacionales de la Salud, proporciona información sobre las investigaciones clínicas. Usted puede ver las investigaciones sobre esta condición en el Clinicaltrials.gov. Use el término “N-Acetylglutamate Synthetase Deficiency” o un término más amplio, “Urea cycle disorders” para ver los estudios disponibles. Recomendamos que comparta esta información con los médicos para que analicen los estudios y determinen la indicación de la participación en algún estudio. (en inglés)
Para obtener información sobre los ensayos clínicos en Europa, póngase en contacto con: Clinicaltrialsregister.eu
Gessler P, Buchal P, Schwenk HU, Wermuth B. Favourable long-term outcome after immediate treatment of neonatal hyperammonemia due to N-acetylglutamate synthase deficiency. Eur J Pediatr. 2010;169:197-199.
Krivitzky L, Babikian T, Lee HS, et al. Intellectual, adaptive, and behavioral functioning in children with urea cycle disorders. Pediatr Res. 2009;66:96-101.
Yudkoff M, Ah Mew N, Payan I, et al. Effects of a single dose of N-carbamylglutamate on the rate of ureagenesis. Mol Genet Metab. 2009;98:325-330.
Tuchman M, Caldovic L, Daikhin Y, et al. N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic academia as measured by isotopic incorporation and blood biomarkers. Pediatr Res. 2008;64:213-217.
Deignan JL, Cederbaum SD, Grody WW. Contrasting features of urea cycle disorders in human patients and knockout mouse models. Mol Genet Metab. 2008;93:7-14.
Caldovic L, Morizono H, Tuchman M. Mutations and polymorphisms in the human N-acetylglutamate synthase (NAGS) gene. Hum Mutat. 2007;28:754-759.
Caldovic L, Morizono H, Daikhin, et al. Restoration of ureagenesis in N-acetylglutamate synthase deficiency by N-carbamylglutamate. J Pediatr. 2004;145:552-554.
Dammers R, Rubio-Gozalbo ME, Robben SG, et al. N-acetyl-glutamate synthetase deficiency or porto-systemic shunt associated encephalopathy? Acta Paediatr. 2002;91(6):729.
Belanger-Quintana A, Martinez-Pardo M, Garcia MJ, et al. Hyperammonaemia as a cause of psychosis in an adolescent. Eur J Pediatr. 2003 Nov;162(11):773-5.
Lee B, et al. Long-term outcome of urea cycle disorders. J Pediatr. 2000;138:S-62-S71.
Plecko B, et al. Partial N-acetylglutamate synthetase deficiency in a 13-year-old girl: diagnosis and response to treatment with N-carbamylglutamate. Eur J Pediatr. 1998;157:996-98.
Guffon N, et al. A new neonatal case of N-acetylglutamate synthase deficiency treated by carbamylglutamate. J Inherit Metab Dis. 1995;18:61-65.
Batshaw ML. Inborn errors of urea synthesis. Ann Neurol. 1994;35:133-41.
Schubiger G, et al. N-acetylglutamate synthetase deficiency: diagnosis, management and follow-up of a rare disorder of ammonia detoxication. Eur J Pediatr. 1991;150:353-56.
Roth KS. N-acetylglutamate synthetase deficiency. Medscape, November 17, 2014. Available at: https://emedicine.medscape.com/article/941090-overview Accessed April 14, 2016.
McKusick VA., ed. Online Mendelian Inheritance in Man (OMIM). Baltimore. MD: The Johns Hopkins University; Entry No:237310; Last Update:02/11/2015. Available at: https://www.ncbi.nlm.nih.gov/omim/237310 Accessed April 14, 2016.
Usted puede aprender más sobre esta enfermedad en los siguientes sitios en la red:
Note que esta información puede ser bastante técnica por lo que recomendamos que la comparta con un profesional de la salud.
En español:
Las siguientes fuentes de información en inglés también pueden ser de utilidad:
Vea también nuestra página en inglés de NORD: N-Acetylglutamate Synthetase Deficiency
Cuando se tiene una enfermedad rara o poco frecuente es muy importante encontrar a un médico que tenga experiencia en el diagnóstico y en el manejo. De forma general, se recomienda que las personas con enfermedades raras busquen ser atendidas en centros médicos universitarios o terciarios ya que es más probable que los médicos que trabajan en estos centros hayan visto casos similares o tengan interés en la investigación, además de que cuentan con equipos de múltiples especialistas que trabajan en conjunto.
NORD tiene una lista de centros de excelencia en enfermedades raras que incluye muchos de los mejores centros médicos y académicos de los Estados Unidos. Recomendamos que los pacientes compartan esta información con sus médicos para que sean referidos al centro más adecuado y conveniente. Esta lista está en expansión.
Para aprender más sobre NORD visite el siguiente enlace: NORD en Español.
Nota: El sitio web de la Organización Nacional de Enfermedades Raras (NORD), sus bases de datos y su contenido tienen derechos de autor de NORD. Ninguna parte del sitio web de NORD, las bases de datos o los contenidos pueden copiarse de ninguna manera, incluidos, entre otros, los siguientes: descarga electrónica, almacenamiento en un sistema de recuperación o redistribución con fines comerciales sin el permiso expreso por escrito de NORD. Sin embargo, por la presente se otorga permiso para imprimir una copia impresa de la información sobre una enfermedad individual para su uso personal, siempre que dicho contenido no se modifique de ninguna manera y el crédito por la fuente (NORD) y el aviso de derechos de autor de NORD estén incluidos en la copia impresa. Cualquier otra reproducción electrónica u otras versiones impresas está estrictamente prohibida.

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD y la Fundación MedicAlert se han asociado en un nuevo programa para brindar protección a pacientes con enfermedades raras en situaciones de emergencia.
Aprende más https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Asegurarse de que los pacientes y los cuidadores estén equipados con las herramientas que necesitan para vivir su mejor vida mientras manejan su condición rara es una parte vital de la misión de NORD.
Aprende más https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/Este programa de asistencia, primero en su tipo, está diseñado para los cuidadores de un niño o adulto diagnosticado con un trastorno raro.
Aprende más https://rarediseases.org/patient-assistance-programs/caregiver-respite/

{kind=link}