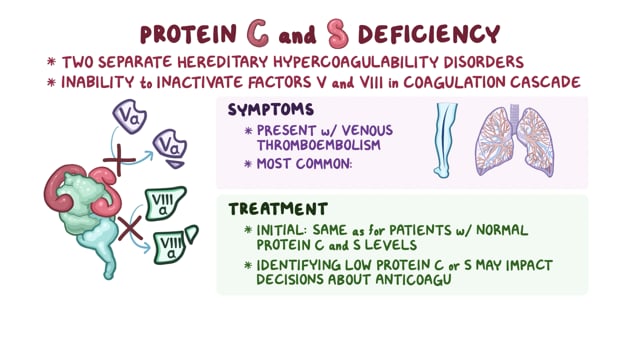
Summary
The spinocerebellar ataxias (SCA) are a group of genetic neurological disorders characterized by difficulty controlling and coordinating voluntary movements (ataxia) due to degenerative changes in the brain and spinal cord (spinocerebellar). Ataxia can lead to an awkward, uncoordinated walk (gait) often accompanied by poor eye-hand coordination, clumsiness, visual problems and abnormal speech (dysarthria).1 The spinocerebellar ataxias are slowly progressive, which means the symptoms usually worsen over time.2 The specific symptoms can vary greatly from one person to another and the age of onset and rate of the progression of these disorders can also vary greatly.2 The spinocerebellar ataxias are inherited in an autosomal dominant pattern. There are no FDA-approved therapies for these disorders. The treatments are symptomatic and supportive and designed to maximize function and reduce complications.3,4
Introduction
The classification of SCAs is complicated and has changed or evolved over time. There are approximately 50 different recognized subtypes of spinocerebellar ataxia.5,6 There are other forms of ataxia that are different from the spinocerebellar ataxias, including other inherited forms (autosomal recessive ataxia, X-linked ataxia, mitochondrial). Ataxia can also occur as a symptom of other genetic disorders including autoimmune disorders or disorders that occur because of cancer (paraneoplastic syndromes).1,4 Ataxia can also be acquired through infections, injuries, or from other external causes.1 Ataxia itself is a general term that is associated with numerous different disorders.
Please complete this form to access the requested resource.