Summary
Pompe disease is a rare disease continuum with variable rates of disease progression and different ages of onset. First symptoms can occur at any age from birth to late adulthood. Earlier onset compared to later onset is usually associated with faster progression and greater disease severity. At all ages, skeletal muscle weakness characterizes the disease, causing mobility problems and affecting the respiratory system.
The most severely affected infants usually present within the first 3 months after birth. They have characteristic heart (cardiac) problems (dysfunction due to heart enlargement) in addition to generalized skeletal muscle weakness and a life expectancy of less than 2 years, if untreated (classic infantile Pompe disease). Less severe forms of Pompe disease with onset during childhood, adolescence, or adulthood, rarely manifest cardiac problems, but gradually lead to walking disability and reduced respiratory function.
The scientific literature has different ways of subdividing the clinical spectrum of Pompe disease. Some articles describe ‘classic infantile’, ‘childhood’ and ‘adult’ Pompe disease while others discuss ‘infantile-onset’ (IOPD) and ‘late-onset’ (LOPD) disease.
Pompe disease is a rare, multisystemic, hereditary disease, which is caused by ‘pathogenic variations’ (abnormalities / mutations) in the ‘GAA gene’.
The GAA gene contains the genetic information for the production and function of a protein called ‘acid alpha-glucosidase’ (GAA). Shortage of this protein hampers the degradation of a complex sugar named ‘glycogen’ into a simple sugar named ‘glucose’. Therefore, glycogen starts to accumulate in all kinds of tissues, but primarily in skeletal muscle, smooth muscle and cardiac muscle, where it causes damage to tissue structure and function.
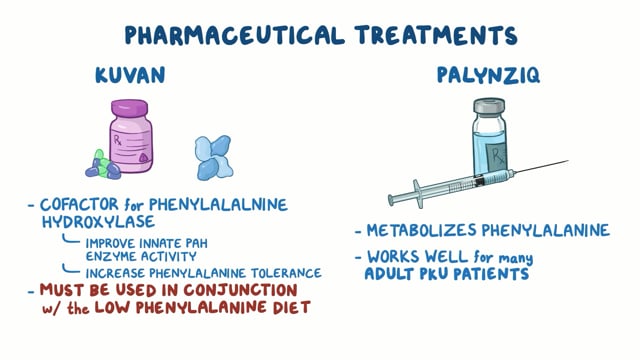
‘Enzyme replacement therapy’ (ERT), the only treatment presently available, aims to replenishing the shortage of GAA by intravenous administration of industrially made ‘rhGAA’ (recombinant human GAA).
Pompe disease is inherited in an autosomal recessive genetic pattern, which implies that healthy parents can have affected children.
Introduction
The human body can be seen as an assembly of interconnecting organs. Organs are composed of organ specific tissues, and tissues are composed of specialized cells like muscle cells, nerve cells, etc. Pompe disease belongs to a group of diseases known as the ‘lysosomal storage disorders’ (LSDs). Lysosomes are small compartments inside the cells wherein all kind of substances are re-cycled. The substances are degraded by the action of digestive enzymes. More than 50 different LSDs are presently known to be caused by the deficiency of one of these enzymes. Acid alpha-glucosidase (GAA) is one such enzyme and is responsible for the lysosomal degradation of glycogen. A shortage or dysfunction of GAA causes glycogen to accumulate within the lysosomes, which subsequently leads to cellular malfunction, cellular damage, tissue damage, and ultimately organ dysfunction. In Pompe disease, the organ dysfunction is mainly manifested by muscle weakness and muscle wasting.
Pompe disease is not only listed as an LSD, but also as one of the 15 presently known ‘glycogen storage disorders’ (GSDs), a group of metabolic disorders characterized by abnormalities in glycogen synthesis and breakdown.
Pompe disease is known under the alternative names ‘glycogen storage disease type II’ (GSDII), acid alpha-glucosidase (GAA) deficiency, and ‘acid maltase’ deficiency (acid maltase is another name for acid alpha-glucosidase).
Terminology
Pompe disease is divided into subtypes: ‘Classic infantile’ refers to the form of Pompe disease that was first described in 1932 and characterized by the onset of symptoms shortly after birth, generalized muscle weakness, and ‘cardiomegaly’ (a far too big heart), in combination with excessive glycogen stored in virtually all organs. Terms like ‘childhood’, ‘juvenile’, and ‘adult’ glycogenosis type II / Pompe disease / Acid maltase deficiency were historically introduced as names for the less severe forms of Pompe disease characterized by delayed onset and usually slower progression. Adult-onset was historically synonymous with ‘late-onset’.
After the introduction of enzyme replacement therapy, the meaning of ‘late-onset’ was increasingly referred to Pompe disease without hypertrophic cardiomyopathy (HCM) (thickened heart muscle).
Currently, the abbreviation IOPD (infantile-onset Pompe disease) refers in most but not all published cases to classic-infantile Pompe disease (some cases of childhood Pompe disease might be included). The abbreviation IPD (infantile Pompe disease) is also used. LOPD (late-onset Pompe disease) refers to all cases in which hypertrophic cardiomyopathy (HCM) did not manifest or was not diagnosed at or under the age of 1 year, as well as to all cases with symptom onset above the age of 1 year.
Please complete this form to access the requested resource.