

Última actualización:
March 21, 2017
Años publicados: 1997, 1999, 2001, 2002, 2017
El síndrome del ojo de gato es un trastorno cromosómico raro que puede ser evidente al nacer. Las personas con este síndrome tienen un cromosoma 22 adicional anormal.
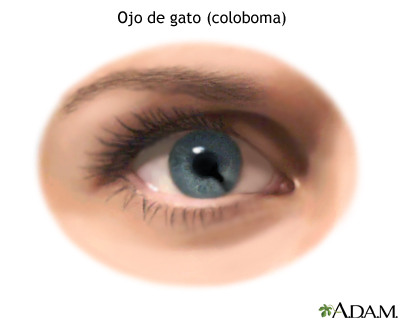
El nombre «síndrome del ojo de gato» se refiere a una anomalía en los ojos, que está presente en más de la mitad de las personas afectadas, y que se conoce como “coloboma”, que parece como una hendidura o espacio en el iris debajo de la pupila lo que resulta en que la pupila alargada de la persona afectada se asemeje al ojo de un gato.
Fuente: MedlinePlus en español. Biblioteca Nacional de la Salud. Síndrome del ojo de gato. 9/12/2021. Disponible en: https://medlineplus.gov/spanish/ency/esp_imagepages/1130.htm
Además del coloboma del ojo, las personas con el síndrome del ojo de gato pueden tener problemas en muchos órganos y sistemas del cuerpo. Estos síntomas resultan del desarrollo anormal durante las etapas embrionaria y fetal. Los síntomas varían, incluso entre miembros de la misma familia por lo que el síndrome del ojo de gato se puede considerar mejor como un espectro de trastornos. Mientras que algunas personas pueden tener pocas o leves manifestaciones, otras pueden tener múltiples malformaciones graves.
NORD agradece a Heather McDermid, PhD, Profesora, Departamento de Ciencias Biológicas, Universidad de Alberta, Edmonton, AB Canadá, por su asistencia en la preparación de este informe en inglés. El informe en inglés fue traducido y modificado por Gioconda Alyea, médica genetista brasileira en mayo del 2023.
Los 3 síntomas clásicos asociados con el síndrome de ojo de gato son:
Sin embargo, estos síntomas son muy variables y se ha estimado que menos de la mitad (como el 41%) de las personas afectadas con el síndrome del ojo de gato tienen estos tres síntomas. En general, las anormalidades asociadas con el síndrome del ojo de gato tienden a involucrar los ojos, los oídos, la región anal, el corazón y/o los riñones, pero otros órganos pueden ser afectados y algunas personas muestran discapacidad intelectual también.
Es posible que algunas personas afectadas no presenten síntomas (sean asintomáticos) o tengan tan pocos síntomas que es posible que no se perciban. Las personas afectadas rara vez tendrán todos los síntomas que se describen a continuación y cada caso es diferente y único. Si un síntoma está presente en un solo caso, eso puede tener una causa independiente (que no forma parte del síndrome). Se recomienda que los padres de una persona afectada hablen con los médicos sobre los posibles síntomas asociados con este síndrome y sobre el pronóstico general.
Algunos estudios han mostrado la frecuencia de las señales y los síntomas del síndrome del ojo de gato. Las características típicas del síndrome del ojo de gato presentes en más del 10 % de las personas afectadas. Según 2 estudios independientes realizados en 2001, las frecuencias de las señales y síntomas en los casos del síndrome de ojo de gato son:
Usted también puede visitar el siguiente enlace de HPO, Ontología del Fenotipo Humano de Orphanet, la base de dados europea de enfermedades raras, que se basa en un análisis de la literatura biomédica para presentar las señales y síntomas por orden de frecuencia de ocurrencia en la población de pacientes, y por orden alfabético dentro de cada grupo de frecuencias. Señales y síntomas HPO.
El síndrome del ojo de gato es un trastorno raro que, por lo general, es causado por la presencia de un fragmento cromosómico adicional, en el que el brazo corto (p) y una pequeña porción del brazo largo (q) del cromosoma 22 suelen estar presentes en cuatro copias (tetrasomía parcial) en lugar de dos copias en las células del cuerpo. También puede ser causado por trisomía parcial del cromosoma 22 y por la triplicación intracromosómica de la región 22q11.
Los cromosomas contienen las informaciones genéticas de cada individuo y se encuentran en el núcleo de todas las células del cuerpo. Los cromosomas vienen en pares. Normalmente, cada célula en el cuerpo humano tiene 23 pares de cromosomas (46 cromosomas en total), de los cuales la mitad proviene de la madre y la otra mitad del padre. Hay 22 pares de cromosomas autosómicos y un par de cromosomas sexuales (el X y el Y) que determinan el género masculino o femenino. Las mujeres tienen 2 cromosomas X (son 46, XX) y los varones tienen un cromosoma X y uno Y (son 46, XY).
Cada cromosoma tiene un brazo corto designado como «p», un brazo largo identificado por la letra «q» y una región estrecha en la que se unen los dos brazos (centrómero). Cuando los cromosomas se estudian con tintas especiales, los cromosomas se subdividen en regiones llamadas “bandas” que se numeran hacia afuera desde el centrómero. Por ejemplo, el brazo corto del cromosoma 22 incluye las bandas 22p11.1 a 22p13; el extremo o “terminal” del brazo corto se conoce como 22pter. El brazo largo incluye las bandas 22q11.1 a 22q13.
Las personas con una composición cromosómica normal tienen dos cromosomas 22, los cuales consisten en un brazo corto (22p), un brazo largo (22q) y un centrómero. La mayoría de las personas afectadas con el síndrome del ojo del gato tienen cromosomas pequeños adicionales que son compuestos por un segmento de la mitad superior (ter) del brazo corto (p) del cromosoma 22 (22pter) y por una pequeña región del brazo largo (q) del cromosoma 22 (22q11).
En la mayoría de los casos del síndrome, el cromosoma 22 adicional, 22pter-22q11, también llamado “cromosoma marcador bisatelitado supernumerario” está presente cuatro veces (tetrasomia) en las células del cuerpo y, en algunos casos, tres veces (trisomía). En como en un tercio de los casos este cromosoma adicional puede estar presente sólo en algunas células del cuerpo y no en otras (mosaicismo). No se puede saber los síntomas específicos o la gravedad de la enfermedad en un bebé con una forma de mosaico del síndrome del ojo de gato.
En raras ocasiones, una porción del segmento cromosómico 22q11 puede aparecer tres veces, una vez en un cromosoma 22 normal y dos veces en un cromosoma 22 con una duplicación interna (triplicación intracromosómica de la región 22q11).
Se cree que hay una porción de la región 22q11 que es responsable por de todas o la mayoría de las características asociadas con el síndrome. Esta región se denomina “región crítica del síndrome del ojo de gato” y contiene aproximadamente 12 genes. Se están realizando investigaciones para aislar y caracterizar los genes responsables por las características asociadas con el síndrome de ojo de gato.
Herencia
En la mayoría de los casos, la anomalía cromosómica parece surgir «de novo» o al azar (sin ser heredada) debido a un error en la división de las células reproductivas de uno de los padres (error meiótico); en tales casos, el padre tiene cromosomas normales. Es importante saber que esto no es culpa de nadie.
En unos pocos casos, el cromosoma marcador puede ser heredado de uno de los padres, que tiene síntomas leves (y que muchas veces no está diagnosticado) o que tiene mosaicismo. Se han informado casos en los que el mosaicismo de esta anomalía cromosómica puede transmitirse a través de varias generaciones en algunas familias; sin embargo, como comentado antes, los síntomas son variados y puede suceder que aquellos con características múltiples o graves sean diagnosticados, mientras que otros casos en la familia no son diagnosticados. En cualquier caso, las personas con este síndrome que tienen hijos corren un riesgo importante de transmitir el cromosoma marcador adicional a sus hijos.
Se necesita más investigación para aprender más sobre los complejos mecanismos cromosómicos y genéticos que pueden causar los problemas y sobre la transmisión del síndrome.
El síndrome del ojo de gato ha sido reconocido por más de un siglo. Se han descrito más de 100 casos en la literatura médica, incluidos casos aparentemente esporádicos y casos familiares. Algunos estudios han mostrado que afecta a una persona en 50 000 o a una persona en 150 000 personas en el noreste de Suiza. Sin embargo, debido a que algunos individuos afectados desarrollan pocas características asociadas, el trastorno puede pasar desapercibido en algunas personas y deben existir muchos más individuos afectados, pero no han sido descritos en la literatura médica. Actualmente no hay forma de saber con seguridad cuantas personas tienen este síndrome.
El diagnóstico del síndrome del ojo de gato se basa en la presencia de las señales y de los síntomas de las personas afectadas y se establece con la identificación de la presencia de material extra derivado del cromosoma 22q11.
Es posible que se sospeche un diagnóstico del síndrome antes del nacimiento (prenatalmente) en base a pruebas especializadas, como ultrasonido, amniocentesis y/o muestreo de vellosidades coriónicas (CVS). Durante la ecografía fetal, las ondas de sonido reflejadas crean una imagen del feto en desarrollo, lo que potencialmente revela ciertos defectos, como un defecto cardíaco, que podría sugerir que el bebé este afectado con alguna enfermedad genética. Durante la amniocentesis, se extrae y analiza una muestra de líquido amniótico que contiene células fetales, mientras que el CVS implica la extracción de muestras de tejido de una parte de la placenta. Los estudios cromosómicos realizados en dichas células pueden revelar el cromosoma anormal adicional característico del síndrome.
El síndrome del ojo de gato puede reconocerse después del nacimiento (posnatalmente) mediante una evaluación clínica que detecta los síntomas característicos, como coloboma, pliegues palpebrales inclinados hacia abajo (fisuras palpebrales), marcas y/o fosas preauriculares, orejas malformadas con ausencia del canal auditivo externo, atresia anal, defectos cardíacos y malformaciones renales. El diagnóstico se confirma mediante estudios cromosómicos estándar para identificar el cromosoma anormal adicional o una duplicación de la región 22q11.
Una vez que se realiza un diagnóstico cromosómico, también se pueden realizar varias pruebas especializadas para determinar qué tipo de problemas relacionados con este síndrome tiene la persona afectada.
Es importante que se haga una evaluación cardíaca completa para detectar cualquier anomalía del corazón que pueda estar presente, incluido un examen clínico completo, evaluación de los sonidos cardíacos y pulmonares mediante el uso de un estetoscopio, estudios de rayos X, electrocardiografía (EKG), ecocardiografía, cateterismo cardíaco y/u otros estudios cardíacos.
Las pruebas adicionales deben incluir un examen ocular completo y un control cuidadoso de la audición. El diagnóstico temprano de una posible discapacidad visual y/o pérdida auditiva puede garantizar una intervención rápida y una corrección o tratamiento de apoyo oportunos y tempranos.
También se pueden usar técnicas de imagen especializadas y/u otras pruebas para detectar y/o caracterizar posibles defectos gastrointestinales, genitourinarios, renales, esqueléticos o biliares, así como otras anomalías físicas que pueden ocurrir en las personas con este síndrome. La investigación de la función cognitiva también puede ser apropiada.
El tratamiento puede requerir los esfuerzos coordinados de un equipo de profesionales médicos, como pediatras, cirujanos, especialistas del corazón (cardiólogos), especialistas del tracto digestivo, especialistas de los ojos; profesionales de la salud que detectan, evalúan y ayudan a manejar los problemas auditivos; médicos que diagnostican y tratan trastornos del esqueleto, músculos, articulaciones y tejidos relacionados (ortopedistas); y/u otros profesionales de la salud.
El manejo de la enfermedad depende de los síntomas específicos que la persona afectada tiene. Los defectos cardíacos congénitos pueden ser tratados con medicamentos o con cirugía. La atresia anal se corrige con cirugía. En algunos casos, el tratamiento recomendado también puede incluir cirugía, para corregir los defectos oculares, las anomalías esqueléticas, los defectos genitales, las hernias, las anomalías de la enfermedad de Hirschsprung, la atresia biliar y/u otras malformaciones asociadas. Las cirugías especificas pueden depender del tamaño, la naturaleza, la gravedad y/o la combinación de anomalías, de los síntomas asociados; de la edad del y de otros factores.
Antes y después de la cirugía para ciertos defectos cardíacos, las personas pueden tener una infección bacteriana del revestimiento y de las válvulas del corazón (endocarditis). Por lo tanto, es importante que se receten antibióticos de forma preventiva (profiláctica) antes y después de ciertos procedimientos quirúrgicos y de las visitas al dentista. Además, las infecciones respiratorias deben tratarse temprano de forma efectiva.
Para las personas con ciertas anomalías esqueléticas, el tratamiento puede incluir fisioterapia y diversas técnicas ortopédicas, que posiblemente incluyan cirugía. Además, las personas con baja estatura severa que presentan deficiencia de la hormona del crecimiento pueden tratarse con hormona del crecimiento.
La intervención temprana es importante para garantizar que los niños con este síndrome alcancen su potencial máximo. Los servicios especiales que pueden ser beneficiosos incluyen educación especial de recuperación, apoyo social especial y/u otros servicios médicos, sociales y/o vocacionales.
El asesoramiento genético también será beneficioso para las personas afectadas y sus familias. Es posible que se recomienden estudios cromosómicos para los padres de las personas afectadas para determinar si portan el cromosoma anormal que causa el síndrome del ojo de gato o si tienen mosaicismo para este cromosoma, en especial, si manifiestan alguna característica que pueda estar asociada con este síndrome. El asesoramiento genético también puede beneficiar a los adultos con este síndrome que estén interesados en tener hijos.
El sitio en la red de Clinical Trials, desarrollado por los Institutos Nacionales de la Salud, proporciona información sobre las investigaciones clínicas. Usted puede ver las investigaciones en el siguiente enlace: Clinicaltrials.gov Recomendamos que comparta esta información con los médicos para que analicen los estudios y determinen la indicación de la participación en algún estudio. (en inglés)
Para obtener información sobre los ensayos clínicos en Europa, póngase en contacto con: Clinicaltrialsregister.eu
Jones KL, Jones MC, del Campo Casanelles. Eds. Cat Eye Syndrome. In: Smith’s Recognizable Patterns of Human Malformation. 7th ed. Elsevier Saunders, Philadelphia, PA; 2013:66-67.
McDermid H, Bamforth JS. Cat Eye Syndrome. In: NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia, PA. 2003:164-165.
Jedraszak G, Receveur A, Andrieux J, et al. Severe psychomotor delay in a severe presentation of cat-eye syndrome. Case Rep Genet. 2015;2014:943905. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4310452/
Sharma D, Murki S, Pratap T, Vasikarla M. Cat eye syndrome. BMJ Case Rep. 2014;2014. https://www.ncbi.nlm.nih.gov/pubmed/24842361
Hofmann AD, Puri P. Association of Hirschsprung’s disease and anorectal malformation: a systemic review. Pediatr Surg Int. 2013;29:913-917. https://www.ncbi.nlm.nih.gov/pubmed/23948812
Melo C, Gama-de-Sousa S, Almeida F, et al. Cat eye syndrome and growth hormone deficiency with pituitary anomalies: a case report and review of the literature. Gene. 2013;529:186-189. https://www.ncbi.nlm.nih.gov/pubmed/23928108
Belangero SI, Pacanaro AN, Bellucco FT, et al. Wide clinical variability in cat eye syndrome patients: four non-related patients and three patients from the same family. Cytogenet Genome Res. 2012:138:5-10. https://www.ncbi.nlm.nih.gov/pubmed/22890013
Knijnenburg J, van Bever Y, Hulsman LOM, et al. A 600 kb triplication in the cat eye syndrome critical region causes anorectal, renal and preauricular anomalies in a three-generation family. Eur J Hum Genet. 2012;20:986-989. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3421127/
Romagna ES, Appel da Silva MC, Ballardin PA. Schmid-Fraccaro syndrome: severe neurologic features. Pediatr Neurol. 2010;42:151-153. https://www.ncbi.nlm.nih.gov/pubmed/20117756
Rosa RF, Mombach R, Zen PR, Graziadio C, Paskulin GA. Clinical characteristics of a sample of patients with cat eye syndrome. Rev Assoc Med Bras. 2010;56:462-465. https://www.ncbi.nlm.nih.gov/pubmed/20835645
Gomez-Lado C, Eiris J, Martinez-Yriarte JM, Blanco O, Castro-Gago M. Duane’s syndrome and 22 marker chromosome: a possible cat-eye syndrome. Acta Pediatr. 2006;95:1510-1511. https://www.ncbi.nlm.nih.gov/pubmed/17062489
Lalani S R, Safiullah A M, Fernbach S D, et al. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am. J. Hum. Genet. 2006; 78: 303-314.
McDermid HE, Morrow BE. Genomic disorders on 22q11. Am H Hum Genet. 2002;70:1077-1088. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC447586/
Rosias PR, Sijstermans JM, Theunissen PM, et al., Phenotypic variability of the cat eye syndrome. Case report and review of the literature. Genet Couns. 2002;12:273-82. https://www.ncbi.nlm.nih.gov/pubmed/11693792
Berends MJ, Tan-Sindhunata G, Leegte B, van Essen AJ. Phenotypic variability of Cat-Eye syndrome. Genet Couns. 2001;12:23-34. https://www.ncbi.nlm.nih.gov/pubmed/11332976
Footz TK, Brinkman-Mills P, Banting GS, et al..Analysis of the cat eye syndrome critical region in humans and the region of conserved synteny in mice: a search for candidate genes at or near the human chromosome 22 pericentromere. Genome Res. 2001;11(6):1053-70.
Edelmann L, Pandita RK, Spiteri E, et al. A common molecular basis for rearrangement disorders on chromosome 22q11. Hum Mol Genet. 1999;8:1157-67. https://www.ncbi.nlm.nih.gov/pubmed/10369860
Masukawa H, Ozaki T, Nogimori T. Cat eye syndrome with hypogonadotropic hypogonadism. Intern Med. 1998;37:853-56. https://www.ncbi.nlm.nih.gov/pubmed/9840707
McTaggart KE, Budarf ML, Driscoll DA, et al. Cat eye syndrome chromosome breakpoint clustering: identification of two intervals also associated with 22q11 deletion syndrome breakpoints. Cytogenet Cell Genet. 1998;81:222-28. https://www.ncbi.nlm.nih.gov/pubmed/9730608
Mears AJ, el-Shanti H, Murray JC, McDermid HE, Patil RS. Minute supernumerary ring chromosome 22 associated with cat eye syndrome: further delineation of the critical region. Am J Hum Genet. 1995;57:667-73. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1801256/
Mears AJ, Duncan AM, Budarf ML, et al. Molecular characterization of the marker chromosome associated with cat eye syndrome. Am J Hum Genet.1994;55:134-42. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1918240/
Tupler R, Hoeller P, Pezzolo A, Maraschio P. Maternal derivation of inv dup (22) and clinical variation in cat-eye syndrome. Ann Genet. 1994;37:153-55. https://www.ncbi.nlm.nih.gov/pubmed/7847799
McDermid HE, Duncan AMV, Brasch KR, et al. Characterization of the supernumerary chromosome in cat eye syndrome. Science. 1986;232: 646-648
Schinzel AW. Schmid et al. The “Cat Eye syndrome”: Dicentric small marker chromosome probably derived from a No. 22 (Tetrasomy 22pter→q11) associated with a characteristic phenotype. Human Genetics 1981;57(2):148-158.
McKusick VA., ed. Online Mendelian Inheritance in Man (OMIM). Baltimore. MD: The Johns Hopkins University; Entry No:115740; Last Update:08/13/2009. Available at: https://omim.org/entry/115470 Accessed March 21, 2017.
Usted puede aprender más sobre el síndrome del ojo de gato en los siguientes sitios en la red:
Cuando se tiene una enfermedad rara o poco frecuente es muy importante encontrar a un médico que tenga experiencia en el diagnóstico y en el manejo. De forma general, se recomienda que las personas con enfermedades raras busquen ser atendidas en centros médicos universitarios o terciarios ya que es más probable que los médicos que trabajan en estos centros hayan visto casos similares o tengan interés en la investigación, además de que cuentan con equipos de múltiples especialistas que trabajan en conjunto.
NORD tiene una lista de centros de excelencia en enfermedades raras que incluye muchos de los mejores centros médicos y académicos de los Estados Unidos. Recomendamos que los pacientes compartan esta información con sus médicos para que sean referidos al centro más adecuado y conveniente. Esta lista está en expansión.
Para aprender más sobre NORD visite el siguiente enlace: NORD en Español.
Nota: El sitio web de la Organización Nacional de Enfermedades Raras (NORD), sus bases de datos y su contenido tienen derechos de autor de NORD. Ninguna parte del sitio web de NORD, las bases de datos o los contenidos pueden copiarse de ninguna manera, incluidos, entre otros, los siguientes: descarga electrónica, almacenamiento en un sistema de recuperación o redistribución con fines comerciales sin el permiso expreso por escrito de NORD. Sin embargo, por la presente se otorga permiso para imprimir una copia impresa de la información sobre una enfermedad individual para su uso personal, siempre que dicho contenido no se modifique de ninguna manera y el crédito por la fuente (NORD) y el aviso de derechos de autor de NORD estén incluidos en la copia impresa. Cualquier otra reproducción electrónica u otras versiones impresas está estrictamente prohibida.

NORD y la Fundación MedicAlert se han asociado en un nuevo programa para brindar protección a pacientes con enfermedades raras en situaciones de emergencia.
Aprende más https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Asegurarse de que los pacientes y los cuidadores estén equipados con las herramientas que necesitan para vivir su mejor vida mientras manejan su condición rara es una parte vital de la misión de NORD.
Aprende más https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/Este programa de asistencia, primero en su tipo, está diseñado para los cuidadores de un niño o adulto diagnosticado con un trastorno raro.
Aprende más https://rarediseases.org/patient-assistance-programs/caregiver-respite/