

Last updated:
October 05, 2021
Years published: 1986, 1987, 1988, 1989, 1991, 1993, 1994, 1996, 1997, 1999, 2003, 2004, 2009, 2021
NORD gratefully acknowledges Monty Worthington, MS, NORD Editorial Intern from the Stanford University MS Program in Human Genetics and Genetic Counseling and MaryAnn Campion, EdD, MS, CGC, Clinical Associate Professor, Director, Stanford University MS Program in Human Genetics and Genetic Counseling, for assistance in the preparation of this report.
Charcot-Marie-Tooth (CMT) disease is a group of disorders in which the motor and/or sensory peripheral nerves are affected, resulting in muscle weakness and atrophy as well as sensory loss. Symptoms occur first in the distal legs and later in the hands. The nerve cells in individuals with this disorder are not able to send electrical signals properly because of abnormalities in the nerve axon or abnormalities in the insulation (myelin) around the axon. In CMT specific gene mutations are responsible for the abnormal function of the peripheral nerves. In many forms of CMT these genes are known and in others, while the condition is known to be inherited, the specific gene has not yet been identified.
Symptoms of CMT disease usually begin gradually in adolescence, but can begin earlier or later. In almost all patients, the longest nerve fibers are affected first. Over time, affected individuals may lose the normal use of their feet, hands, legs and arms. Common early signs and symptoms can include decreased sensitivity to heat, touch or pain, muscle weakness in the hand, foot or lower leg, trouble with fine motor skills, high-stepped gait (foot drop), loss of muscle mass in the lower leg, frequent tripping or falling, hammertoe, high foot arch and flat feet. Stretch reflexes may be lost. The disease is slowly progressive and variable, and those affected may remain active for years and live a normal life span. In the most severe cases, breathing difficulties can hasten death.
CMT is known to be a genetic condition caused by genes that have abnormal changes affecting their function. There are now over 100 genes that are known to be responsible for various forms of CMT. A single gene, PMP22, when duplicated, is the cause of around 50% of cases of CMT, while damaging changes in some genes that cause CMT can be extremely rare and found only in a few families. In around 40% of cases of CMT, no responsible gene has yet been identified.
Genetic diseases are caused by changes in a single gene or in a combination of genes for a particular trait that are on the chromosomes received from the father and the mother. At this time, genetic causes for CMT can only be identified when a single gene is responsible for the condition, but not when the effect of several genes with damaging changes, known as polygenic inheritance, is suspected. Single genes that cause CMT can be inherited in an autosomal dominant, autosomal recessive, X-linked or X-linked dominant pattern.
Dominant genetic disorders occur when only a single copy of a non-working gene is necessary to cause a particular disease. The non-working gene can be inherited from either parent or can be the result of a changed (mutated) gene in the affected individual. The risk of passing the non-working gene from an affected parent to an offspring is 50% for each pregnancy. The risk is the same for males and females.
Recessive genetic disorders occur when an individual inherits a non-working gene from each parent. If an individual receives one working gene and one non-working gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the non-working gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier, like the parents, is 50% with each pregnancy. The chance for a child to receive working genes from both parents is 25%. The risk is the same for males and females.
X-linked genetic disorders are conditions caused by a non-working gene on the X chromosome and manifest mostly in males. Females that have a non-working gene present on one of their X chromosomes are carriers for that disorder. Carrier females usually do not display symptoms because females have two X chromosomes and only one carries the non-working gene. Males have one X chromosome that is inherited from their mother and if a male inherits an X chromosome that contains a non-working gene he will develop the disease. Female carriers of an X-linked disorder have a 25% chance with each pregnancy to have a carrier daughter like themselves, a 25% chance to have a non-carrier daughter, a 25% chance to have a son affected with the disease and a 25% chance to have an unaffected son. If a male with an X-linked disorder is able to reproduce, he will pass the non-working gene to all of his daughters who will be carriers. A male cannot pass an X-linked gene to his sons because males always pass their Y chromosome instead of their X chromosome to male offspring.
X-linked dominant disorders are caused by a non-working gene on the X chromosome and occur mostly in females. Females with these rare conditions are affected when they have an X chromosome with the non-working gene for a particular disease. Males with a non-working gene for an X-linked dominant disorder are more severely affected than females and often do not survive.
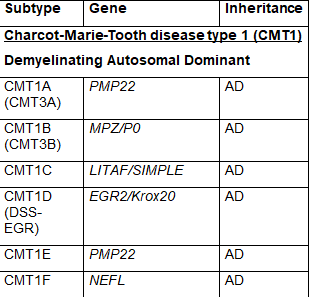
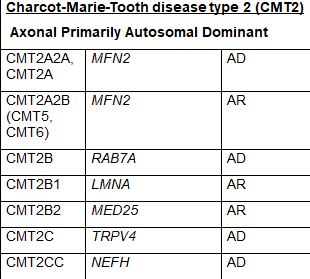
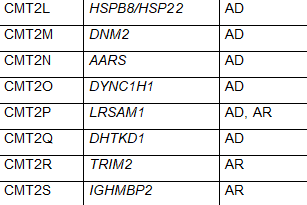
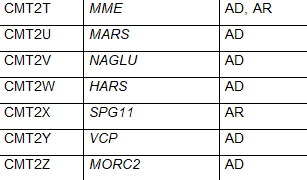
The common naming system for CMT disease is a subdivision into several major types termed CMT1, CMT2, CMT3, CMT4, CMTX, CMTDI and CMTRI. If a responsible gene has been identified, a letter is added to the name, such as CMT1A, CMT1B etc. Recently, new naming schemes have been proposed to simplify this by basing names on inheritance pattern, whether the cause is demyelinating, axonal or intermediate and if the causative gene if known. Table 1 lists CMT types using the common nomenclature and lists causative genes. Some of the types were named before genetic causes were well understood, so there is some redundancy in this naming system and some types have proven to be inaccurately attributed to a specific gene making certain subtype names obsolete. For these reasons some names are no longer in common use.
CMT1 is the most common form of CMT and is characterized by abnormalities in nerve myelin that leads to decreased nerve conduction velocities. Peripheral nerve myelin is formed by Schwann cells, and abnormal changes in genes involved in formation and function of these Schwann cells leads to demyelination. CMT1 is inherited in an autosomal dominant pattern. CMT1 has been further subdivided into subtypes from CMT1A – 1F based on specific gene abnormalities. CMT1A is by far the most common single subtype of CMT accounting for around 50% of all cases of CMT and is caused by a duplication of the PMP22 gene. Other subtypes of CMT1 and responsible genes are listed in Table 1.
CMT2 is a form of the condition in which nerve conduction velocities are usually normal or slightly slower than normal but nerve signal strength is reduced. CMT2 is caused by abnormal genes involved in the structure and function of axons. CMT2 includes primarily autosomal dominant inheritance patterns. CMT2 has been further subdivided into CMT2A – 2Z based on mutations in specific genes. CMT2A is the most common and is caused by mutations in the MFN2 gene. Other subtypes of CMT2 and responsible genes are listed in Table 1. While CMT2 is meant to indicate autosomal dominant inheritance patterns, some subtypes feature autosomal recessive inheritance.
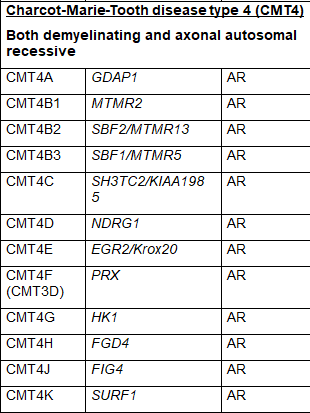
CMT4 is a classification strictly for autosomal recessive forms of CMT. CMT4A is caused by an abnormality in the GDAP1 gene; however the GDAP1 gene is also responsible for CMT2H and CMT2K when the damaging changes to it are sufficient to be transmitted through an autosomal dominant inheritance pattern. Other subtypes of CMT4 and responsible genes are listed in Table 1. SORD-related CMT is the current name for an autosomal recessive axonal form of CMT caused by changes in the SORD gene. This gene was only recently found to be responsible for CMT but may be the most common genetic cause of autosomal recessive CMT. It has not yet been given a classification in the common naming system.
CMTX is an X-linked dominant form of the condition caused by abnormalities in genes located on the X chromosome. CMTX1 (aka CMT1X), caused by changes in the GJB1 gene, accounts for approximately 90% of CMTX. Other subtypes of CMTX and responsible genes are listed in Table 1. Because GJB1 and these other known and unknown genes that cause CMTX are located on the X-chromosome, CMTX primarily affects males, however in CMTX1 and CMTX6 feature an X-linked dominant inheritance with males being more severely affected than females. In CMTX1 and CMTX6, affected females usually have a later onset than males and a milder condition at every age, or may even be asymptomatic, probably due to X inactivation in the myelinating Schwann cells.
Intermediate CMTs include those with “intermediate” conduction velocities and thus an uncertainty regarding whether the neuropathy is primarily axonal or demyelinating. There are both dominant (CMTDI) and recessive (CMTRI) forms of Intermediate CMT. Known causative genes for the subtypes of CMTDI and CMTRI are included in Table 1.
Hereditary neuropathy with liability to pressure palsies (HNPP) is another condition in the CMT group of conditions. HNPP is inherited in an autosomal dominant pattern. Like CMT1A, HNPP is caused by changes to the PMP22 gene. Unlike CMT1A, where the PMP22 gene is usually duplicated, in HNPP one of the PMP22 genes is deleted, so instead of having two copies of PMP22 (one from each parent), HNPP patients only have one copy. This means that patients with HNPP do not produce enough of the PMP22 proteins.
CMT3, also called Dejerine-Sottas disease, is no longer a commonly used name because individuals with this condition have been found to have a gene mutation in one of the genes responsible for CMT1A, CMT1B, CMT1D or CMT4. CMT5 and CMT6 are also historically used terms now attributed to damaging changes in the MFN2 gene that is now known to cause CMT2A2A. Historical names for subtypes that are no longer in use are included in parenthesis next to the contemporary subtype name in Table1.
Hereditary sensory neuropathies (HSN or HSAN) are sometimes included in the CMT group of conditions. Some forms of HSN are related to or identical to forms of CMT and there is overlap with genes responsible for these conditions. Hereditary motor neuropathies (dHMN) are similar to CMT and some experts consider these conditions to be in the same group.
Table 1 Subtypes of CMT, causative genes and inheritance pattern (adapted from Nam,Choi, 2019)



CMT is found worldwide in people of all races and ethnic groups. Prevalence rate estimates from epidemiological studies are highly variable due in part to the wide variation of clinical symptoms and different disease forms as well as discrepancies in what conditions are included within CMT. Recent prevalence estimates range from 9 to 28 per 100,000. Symptoms of CMT usually begin gradually in adolescence, early adulthood or middle age.
The diagnosis of CMT disease can be challenging. Diagnosis is based on physical symptoms, family history and clinical tests. Clinical tests include nerve conduction velocity (NCV) which measures the speed at which impulses travel along the nerves and electromyogram (EMG) which records the electrical activity of muscle cell. With recent advances in molecular genetic testing using both deletion duplication analysis and next generation sequencing (NGS) for patients with a clinical diagnosis of CMT, a genetic cause can be found in about 60% of patients. For CMT1 a genetic cause can be identified in 56-92% of patients. For axonal CMT, genetic causes can be found 17-44% of the time. Panels to test multiple genes can include from 7 to 150 genes, while abnormalities in 4 genes, (PMP22 duplication, GJB1, MFN2, and MPZ) account for about 90% of these genetic diagnoses.
Treatment
Treatment of CMT disease is symptomatic and supportive. A cure is not available so it is important to minimize or stall the symptoms. Comprehensive treatments include physical therapy, shoe orthotics, leg braces and surgery to correct deformities. Complementary therapies may help psychologically, relieve pain and discomfort, and improve overall quality of life. Vocational counseling, anticipating progression of the disorder, may be useful for young patients. Genetic counseling can be helpful with many aspects including genetic diagnosis, connecting with research and support groups and in understanding of recurrence risk.
The Charcot Marie Tooth Association (CMTA) supports the Strategy to Accelerate Research (STAR) program which promotes research including early preclinical investigation of treatments and clinical trials of promising treatments. A list of treatments and trials under investigation is available on their website:
https://www.cmtausa.org/our-research/for-patients-and-families/our-approach/
The CMT Research Foundation (CMTRF) maintains a list of research and clinical trials here: https://cmtrf.org/research/cmt-research-pipeline/
There are many investigational therapies in preclinical or phase 1-3 trials underway. These include trials of therapeutic agents as well as diverse gene therapy approaches. Information on current clinical trials is posted on the internet at www.clinicaltrials.gov
All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Tollfree: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website:
https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, contact:
www.centerwatch.com
For more information about clinical trials conducted in Europe, contact: https://www.clinicaltrialsregister.eu/
JOURNAL ARTICLES
Nam SH, Choi BO/ Clinical and genetic aspects of Charcot-Marie-Tooth disease subtypes. Precision and Future Medicine 2019; 3(2): 43-68.
Vaeth S, Vaeth M, Andersen H, Christensen R, Jensen UB. Charcot-Marie-Tooth disease in Denmark: a nationwide register-based study of mortality, prevalence and incidence. BMJ Open 2017; 7(11): e018048.
Barreto LCLS, Oliveira FS, Nunes PS de França Costa IMP, Garcez CA, Goes GM, et al. Epidemiologic study of Charcot-Marie-Tooth disease: a systematic review. Neuroepidemiology 2016; 46(3):157-165.
Wang Y,Yin F. A review of X-linked Charcot-Marie-Tooth disease. Journal of Child Neurology 2016; 31(6): 761-772.
Foley C, Schofield I, Eglon G, Bailey G, Chinnery PF, Horvath R.Charcot–Marie–Tooth disease in Northern England. Journal of Neurology, Neurosurgery & Psychiatry 2012. 83(5): 572-573.
INTERNET
Bird TD. Charcot-Marie-Tooth (CMT) Hereditary Neuropathy Overview. 1998 Sep 28 [Updated 2021 May 20]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1358/ Accessed June 29, 2021.

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportOnline Mendelian Inheritance In Man (OMIM) has a summary of published research about this condition and includes references from the medical literature. The summary contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. OMIM is authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine.
View reportPlease complete this form to access the requested resource.

