

Last updated:
3/13/2024
Years published: 1987, 1988, 1990, 1995, 1997, 2002, 2007, 2024
NORD gratefully acknowledges Calvin Hawe and Kirsten Tunink, NORD Editorial Interns. University of Notre Dame and David Wenger, PhD, Professor Emeritus, Thomas Jefferson University, for assistance in the preparation of this report.
Krabbe disease belongs to a group of disorders called leukodystrophies, rare genetic disorders that affect the white matter of the brain. Krabbe disease is characterized by a deficiency in the enzyme galactocerebrosidase (GALC) which is an enzyme that uses water molecules to break down certain fats (lipids). Failure to break down these lipids results in deterioration of the protective covering (myelin sheath) surrounding nerves in the brain (demyelination). Characteristic globoid (sphere shaped) cells appear in affected areas of the brain. These enlarged fatty cells accumulate in affected areas and cause a variety of progressive neurological symptoms. Depending on the age that symptoms begin, individuals may present with intellectual and behavioral delays, blindness, deafness, paralysis of facial muscles (pseudobulbar palsy), muscle spasms and seizures. Most patients are diagnosed by measuring GALC enzyme activity in leukocytes taken from a blood sample. Very low activity could indicate a diagnosis of Krabbe disease. In some states, diagnosis may occur through newborn screening. If newborns are diagnosed before 14 days old, they are candidates for early treatment with hematopoietic stem cell transplantation (HSCT). Krabbe disease is inherited in an autosomal recessive pattern.
Krabbe disease has four subtypes based on age of onset: early infantile-onset (<13 months); late-infantile onset (13-36 months), adolescent onset and adult onset. Eighty-five percent (85%) of all patients with Krabbe disease have the early infantile onset type. Early infantile-onset Krabbe disease is marked by initial normal development followed by rapid and severe deterioration. The average age of death after symptoms begin is two years, but it can range between eight months and nine years. Later onset Krabbe disease results in a slower decline in neurologic function and an increased life expectancy averaging eight years after symptoms begin. Adults diagnosed with Krabbe disease can have significant neurologic impairment but can live 30-50 years after diagnosis.
The specific symptoms and severity of Krabbe disease vary from person to person.
Infantile-onset specific symptoms include:
Later-onset symptoms typically include:
In addition, degeneration of certain parts of the brain may cause:
Individuals with Krabbe disease may also have difficulty swallowing (dysphagia) and peripheral neuropathy, a condition characterized by muscle weakness; pain; numbness; redness; and burning or tingling sensations in the affected areas, especially the arms and legs (extremities). Pathological changes may be found on a brain biopsy as well as a decrease in white matter viewed on a magnetic resonance imaging (MRI) test.
Cells in the brain called microglia that normally clean up the excess galactolipids (fatty cells) are not working due to the lack of GALC enzyme. This process transforms them into abnormal cells that have more than one nucleus (multinucleated) and are sphere-shaped (globoid). This affects the protective myelin sheath around the nerves of the brain. This often progresses to cause life-threatening complications.
Krabbe disease is an autosomal recessive genetic disorder caused by changes called pathogenic variants in the GALC gene. The gene variants lead to a deficiency in the enzyme galactosylceramidase. This enzyme metabolizes galactosylceramide, a component of the fatty covering (sheath) around the nerves (myelin). Galactosylceramide metabolism is vital to the destruction of old membranes and the creation of new, healthy myelin membranes, so the buildup of certain galactolipids is toxic to myelin-forming cells.
Myelin ensures a rapid transmission of nerve impulses. When the myelin sheath decays or breaks down, this leads to the loss of neurological function. The enzyme GALC also catalyzes the breakdown of psychosine/galactosylsphingosine, a highly toxic but natural fatty (lipid) byproduct of myelin formation. When psychosine accumulates in the myelinating cells of the central nervous system (oligodendrocytes) and Schwann cells in the peripheral nervous system, then destruction of the myelin (demyelination) occurs.
Recessive genetic disorders occur when an individual inherits a disease-causing gene variant from each parent. If an individual receives one normal gene and one variant gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the gene variant and have an affected child is 25% with each pregnancy. The risk of having a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
Hundreds of disease-causing variants in the GALC gene have been reported. Variants that result in very little or no GALC activity can cause a child to be born with infantile Krabbe disease. Variants that allow some small amount of GALC activity to be produced can cause a person to have a milder form of Krabbe disease.
An estimated 1 in 100,000 newborn babies have Krabbe disease. The more severe infantile-onset disease occurs in 85-90% of patients. The other 10-15% of patients are diagnosed with late-onset disease, though this number may now be higher based on data from state newborn screening programs.
Krabbe disease can be diagnosed by testing the activity of the GALC enzyme in white blood cells (leukocytes) taken from a blood sample, skin cells (fibroblasts) or amniotic fluid cells (through amniocentesis). Some states have newborn screening programs that include a test to measure GALC activity. If newborn screening test results are positive, physicians should follow guidelines for next steps as outlined in the American College of Medical Genetics ACT sheet. In a newborn screening test, GALC activity is measured using liquid chromatography tandem mass spectrometry (LC-MS/MS). Additional measurement of GALC activity can be done using leukocytes taken from a small blood sample from the baby.
Test results that show abnormal GALC activity should be followed up by additional testing including measurement of GALC activity in leukocytes from a small blood sample, measurement of the psychosine concentration in the dried blood spot from newborn screening or red blood cells and possibly sequence analysis of the GALC gene. The GALC 30-kb deletion is a common gene variant in individuals with Krabbe disease, but hundreds of other disease-causing variants gave been identified.
There is a wide range of GALC activity measured in the normal population. This is due to variants in the GALC gene that lower activity but are not disease causing. For this reason, measurement of the psychosine concentration is helpful in accurately identifying patients with Krabbe disease. Elevated psychosine levels (>10nmol/L) strongly support a diagnosis of infantile Krabbe Disease. A psychosine concentration less than 2nmol/L indicates the individual does not have Krabbe disease. Values between 2 and 10 may indicate a later-onset form of Krabbe disease.
Elevated dried blood spot psychosine concentration is an indication of Krabbe disease and it is used as a second–tier newborn screening test when GALC activity is low. Genetic testing in utero for high-risk couples can lead to a diagnosis before birth aiding in early intervention and more successful treatment.
When Krabbe disease is suspected, an assessment for neurological affects will often be performed in addition to genetic testing. This may include a brain magnetic resonance imaging (MRI) test, electroencephalogram (EEG), lumbar puncture for cerebrospinal fluid protein evaluation and a clinical exam. Increased cerebrospinal fluid protein (>72mg/dL) is commonly seen in babies with infantile Krabbe disease. MRI is especially useful when assessing disease progression and determining effectiveness of treatment. An EEG may show abnormal electrical brain waves.
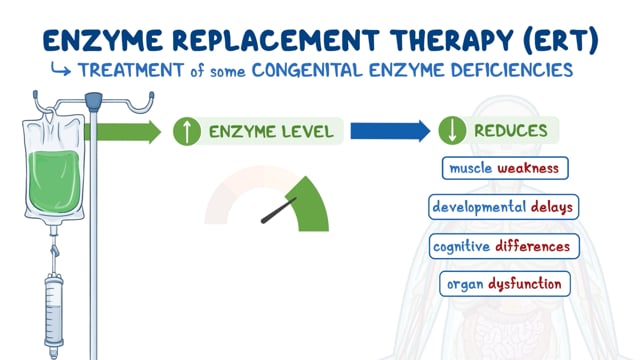
Hematopoietic stem cell transplantation (HSCT) is the current standard treatment for Krabbe disease. This is stem-cell transplant typically from bone marrow or the umbilical cord. Asymptomatic individuals benefit most from this treatment, initially showing near-normal development with some motor delays. The outcomes of this treatment for symptomatic patients are variable and based on age and progression of disease and symptoms. HSCT is usually not recommended for symptomatic individuals. HSCT effectiveness can be monitored with psychosine concentration, which is a biomarker for treatment with HSCT and should decrease with successful therapy. HSCT slows but does not reverse the progression of Krabbe disease, even when started in presymptomatic children.
When HSCT is not an option or is unsuccessful, treatment is symptomatic and supportive. Routine examination and testing may help doctors understand the progression of the disease and to determine the best treatment approach. Numerous specialists – including physical and occupational therapists – may be needed for the successful management of symptoms. Reports based on data from several natural history studies of untreated patients have been published.
It has been suggested that patients avoid antipsychotics, multiple seizure medications and routine childhood vaccinations as these may accelerate neurodegeneration and disease progression.
Genetic counseling is recommended for families of patients with Krabbe disease.
Gene therapy combined with HSCT is being studied in a clinical trial for infants diagnosed with Krabbe disease.
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Tollfree: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website: https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, contact: www.centerwatch.com
For information about clinical trials conducted in Europe, contact: https://www.clinicaltrialsregister.eu/
JOURNAL ARTICLES
Hatton C, Ghanem SS, Koss DJ, et al. Prion-like α-synuclein pathology in the brain of infants with Krabbe disease. Brain. 2022;145(4):1257-1263. doi:10.1093/brain/awac002
LeVine SM, Tsau S. Substrate reduction therapy for krabbe disease: Exploring the repurposing of the antibiotic D-cycloserine. Front Pediatr. 2022;9:807973. doi: 10.3389/fped.2021.807973 [doi].
Lacono S, Del Giudice E, Leon A, La Bella V, Spataro R. A novel compound heterozygous mutation in GALC associated with adult-onset krabbe disease: Case report and literature review. Neurogenetics. 2022. doi: 10.1007/s10048-021-00682-1 [doi].
Nasir G, Chopra R, Elwood F, Ahmed SS. Krabbe disease: Prospects of finding a cure using AAV gene therapy. Front Med (Lausanne). 2021;8:760236. doi: 10.3389/fmed.2021.760236 [doi].
Cachón-González MB, Wang S, Cox TM. Expression of Ripk1 and DAM genes correlates with severity and progression of krabbe disease. Hum Mol Genet. 2021;30(22):2082-2099. doi: 10.1093/hmg/ddab159 [doi].
Zhang X, Niu G, Song P, et al. Compound heterozygous pathogenic variants in the GALC gene cause infant-onset krabbe disease. Transl Pediatr. 2021;10(10):2552-2562. doi: 10.21037/tp-21-403 [doi].
Wenger DA, Luzi P, Rafi MA. Advances in the diagnosis and treatment of krabbe disease. Int J Neonatal Screen. 2021;7(3):57. doi: 10.3390/ijns7030057. doi: 10.3390/ijns7030057 [doi].
Liao HC, Jack R, Scott AI. Galactocerebrosidase activity by liquid-chromatography tandem mass spectrometry for clinical diagnosis of krabbe disease. Clin Chim Acta. 2021;519:300-305. doi: S0009-8981(21)00159-5 [pii].
Babcock MC, Mikulka CR, Wang B, et al. Substrate reduction therapy for krabbe disease and metachromatic leukodystrophy using a novel ceramide galactosyltransferase inhibitor. Sci Rep. 2021;11(1):14486-1. doi: 10.1038/s41598-021-93601-1 [doi].
Basheeruddin K, Shao R, Balster F, Gardley P, Ashbaugh L. Newborn screening for krabbe disease-illinois experience: Role of psychosine in diagnosis of the disease. Int J Neonatal Screen. 2021;7(2):24. doi: 10.3390/ijns7020024. doi: 10.3390/ijns7020024 [doi].
Bradbury AM, Bongarzone ER, Sands MS. Krabbe disease: New hope for an old disease. Neurosci Lett. 2021;752:135841. doi: S0304-3940(21)00219-6 [pii].
Fukazawa R, Takeuchi H, Oka N, Shibuya T, Sakai N, Fujii A. Adult krabbe disease that was successfully treated with intravenous immunoglobulin. Intern Med. 2021;60(8):1283-1286. doi: 10.2169/internalmedicine.6094-20 [doi].
Yoon IC, Bascou NA, Poe MD, Szabolcs P, Escolar ML. Long-term neurodevelopmental outcomes of hematopoietic stem cell transplantation for late-infantile krabbe disease. Blood. 2021;137(13):1719-1730. doi: 10.1182/blood.2020005477 [doi].
Weinstock NI, Shin D, Dhimal N, et al. Macrophages expressing GALC improve peripheral krabbe disease by a mechanism independent of cross-correction. Neuron. 2020;107(1):65-81.e9. doi: S0896-6273(20)30238-5 [pii].
Beltran-Quintero ML, Bascou NA, Poe MD, et al. Early progression of Krabbe disease in patients with symptom onset between 0 and 5 months. Orphanet J Rare Dis. 2019;14(1):46. Published 2019 Feb 18. doi:10.1186/s13023-019-1018-4
Scott-Hewitt NJ, Folts CJ, Hogestyn JM, Piester G, Mayer-Pröschel M, Noble MD. Heterozygote galactocerebrosidase (GALC) mutants have reduced remyelination and impaired myelin debris clearance following demyelinating injury. Hum Mol Genet. 2017;26(15):2825-2837. doi: 10.1093/hmg/ddx153 [doi].
Escolar ML, Kiely BT, Shawgo E, et al. Psychosine, a marker of krabbe phenotype and treatment effect. Mol Genet Metab. 2017;121(3):271-278. doi: S1096-7192(17)30205-6 [pii].
Nicaise AM, Bongarzone ER, Crocker SJ. A microglial hypothesis of globoid cell leukodystrophy pathology. J Neurosci Res. 2016;94(11):1049-1061. doi: 10.1002/jnr.23773 [doi].
INTERNET
Jain M, De Jesus O. Krabbe Disease. [Updated 2023 Aug 23]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK562315/ Accessed Feb 26, 2024.
Orsini JJ, Escolar ML, Wasserstein MP, et al. Krabbe Disease. 2000 Jun 19 [Updated 2018 Oct 11]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1238/ Accessed Feb 26, 2024.
Krabbe disease. Genetic and Rare Diseases Information Center Website. https://rarediseases.info.nih.gov/diseases/6844/krabbe-disease Accessed Feb 26, 2024.

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportOnline Mendelian Inheritance In Man (OMIM) has a summary of published research about this condition and includes references from the medical literature. The summary contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. OMIM is authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine.
View reportGeneReviews has an article on this condition covering diagnosis, management, and inheritance. Each article is written by one or more experts on the specific disease and is reviewed by other specialists. The article contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. The GeneReviews database is managed by the University of Washington.
View reportMedlinePlus has information about this condition that may include a description, frequency, causes, inheritance, and links to more information. The information is written for the public, including patients, caregivers and families. MedlinePlus is a service of the National Library of Medicine (NLM), which is part of the National Institutes of Health (NIH).
View reportPlease complete this form to access the requested resource.
