

Last updated:
April 17, 2020
Years published: 1988, 1989, 2000, 2002, 2007, 2020
NORD gratefully acknowledges Etienne Leveille, MD Candidate, McGill University School of Medicine, and Oleg A Shchelochkov, MD and Charles Venditti, MD, PhD, National Human Genome Research Institute, National Institutes of Health, for assistance in the preparation of this report.
Summary
Propionic acidemia is a rare metabolic disorder affecting from 1/20,000 to 1/250,000 individuals in various regions of the world. It is characterized by deficiency of propionyl-CoA carboxylase, an enzyme involved in the breakdown (catabolism) of the chemical “building blocks” (amino acids) of proteins. Symptoms most commonly become apparent during the first weeks of life and may include abnormally diminished muscle tone (hypotonia), poor feeding, vomiting, listlessness (lethargy), dehydration and seizures. Without appropriate treatment, coma and death may result. Rarely, the condition may become apparent later in life and may be associated with less severe symptoms and findings. Propionic acidemia is inherited in an autosomal recessive pattern. Individuals with this condition have to follow a specific diet including a low protein intake and specific food formulas (medical foods). Liver transplant is a surgical option that can help decrease the frequency of acute metabolic episodes (decompensation).
Symptoms most commonly develop during the first weeks of life and may include vomiting, listlessness (lethargy), low muscle tone (hypotonia), failure to grow and gain weight at the expected rate (failure to thrive), and dehydration. Approximately 30% of affected infants may also develop seizures. The recurrence or worsening of symptoms may be associated with an infection, constipation, or consumption of high amounts of protein. In some affected infants, episodes of symptoms may alternate with periods of apparently normal health and development.
Without appropriate treatment, episodes of vomiting, lethargy, dehydration, and accumulation of excessive levels of acids in the blood and bodily tissues (acidosis) may lead to coma and death [1-4].
As patients age, they can experience various symptoms affecting nearly all organ systems. These symptoms include brain damage (encephalopathy), hypotonia, intellectual disability, severe vision problems, inflammation of the pancreas (pancreatitis), recurrent vomiting, chronic renal failure, heart failure (cardiomyopathy), heart rhythm problems (prolonged QTc interval) , and osteoporosis which can lead to fractures. Affected individuals can also have a reduced number of cells in their blood, such as reduced red blood cells (anemia), reduced white blood cells (leukopenia), reduced platelets (thrombocytopenia), or a reduced number of all cell types (pancytopenia). These blood abnormalities can cause various symptoms, such as immune deficiency or bleeding problems. Individuals with propionic acidemia are also at an increased risk of having a stroke as early as a few weeks of age [4-10].
Less commonly, propionic acidemia may become apparent in childhood or later in life [11-13]. These affected individuals may not experience sudden, acute episodes of acidosis and tend to come to medical attention due to neurological symptoms such as intellectual disability or cardiomyopathy.
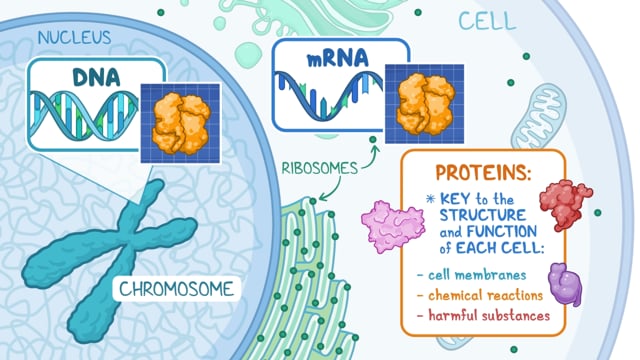
Propionic acidemia is caused by changes (mutations) in the PCCA and PCCB genes resulting in a deficiency of the enzyme propionyl-CoA carboxylase. This enzyme is required for the proper breakdown of the amino acids isoleucine, valine, threonine, and methionine. These amino acids are needed for proper growth and development. Propionyl-CoA carboxylase is also involved in the breakdown of cholesterol, certain fatty acids, and other substances (metabolites) necessary for metabolic actions or processes. Propionyl-CoA carboxylase deficiency leads to accumulation of toxic chemicals (metabolites). Some of these accumulated chemicals harm the mitochondria (power houses inside cells responsible for energy production). [16-20].
Propionic acidemia is inherited in an autosomal recessive pattern.Recessive genetic disorders occur when an individual inherits an abnormal gene from each parent. If an individual receives one normal gene and one abnormal gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the abnormal gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier, like the parents, is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
Propionic acidemia affects males and females in equal numbers. The prevalence varies among different populations and regions. It affects around 1/100,000 to 1/250,000 individuals in most regions of the world. In the Middle East, where there is a high rate of marriage between blood relatives (consanguineous), around 1/20,000 to 1/45,000 individuals are affected. There are around 1/5,000 to ½,000 individuals with this disease in some Saudi tribes. The highest reported rate of propionic acidemia, with 1/1,000 people, has been reported in Greenlandic Inuits [21-29].
Propionic acidemia can be identified at birth through expanded newborn screening by measuring levels of certain metabolites such a propionylcarnitine and methionine in a blood sample. [9, 30].
Most infants with propionic acidemia are diagnosed in the first weeks of life based upon a thorough clinical evaluation, a detailed patient and family history, and molecular genetic testing.
Propionic acidemia may be diagnosed before birth (prenatally) by screening fetal DNA for disease-causing (pathogenic) mutations in the PCCA and PCCB genes. The diagnosis can also be made by measuring the concentration of characteristic metabolites in amniotic fluid or the activity of the propionyl-CoA carboxylase enzyme in fluid or tissue samples obtained from the fetus or uterus during pregnancy (amniocentesis or chorionic villus sampling [CVS]). During amniocentesis, a sample of fluid surrounding the developing fetus is removed and analyzed. CVS involves the removal and examination of tissue from a portion of the placenta.
Treatment
During acute episodes, the treatment of infants with propionic acidemia may require fluid therapy; measures to provide appropriate nutritional intake (e.g., intravenous glucose, with and without intravenous lipids); administration of certain medications to prevent or treat bacterial infection; and other supportive measures as required. In infants with severe disease (e.g., severe acidosis, hyperammonemia), treatment may require procedures that remove excess waste products from the blood (hemodialysis). During hemodialysis, waste products are removed by filtering the blood through an artificial kidney machine. Peritoneal dialysis is a technique during which the peritoneum is used as a natural filtering membrane. (The peritoneum is the two-layered membrane that lines the abdominal wall and covers abdominal organs.). Injection of intravenous bicarbonate can also help reduce the acid load in the body. In addition, until the diagnosis is confirmed, physicians may completely eliminate protein from the newborn’s diet and may also administer biotin, a B complex vitamin that plays a role in the metabolism of certain fatty acids and amino acids.
Long-term treatment includes administration of a low-protein diet, possibly in combination with medical formula (medical foods) that are low in certain amino acids (i.e., amino acids which give rise to propionate, e.g., isoleucine, valine, threonine, and methionine). Infants and children with the disorder may develop secondary deficiency of carnitine, a substance that plays a role in metabolism and the proper use of fatty acids. In such cases, therapy includes administration of L-carnitine (carnitine or levocarnitine). Antibiotic therapy with metronidazole can reduce the burden of propionyl-CoA in the body, as this chemical is produced by some bacteria during fermentation of carbohydrates in our gut.) [4,9,31].
Liver transplant is a potential surgical option for individuals with severe symptoms and frequent recurrent acute episodes (decompensation). Liver recipients generally have a lower risk of decompensation and hospitalization. However, lifelong immunosuppressive therapy is necessary to prevent organ rejection [32-34].
All patients with propionic academia must be followed by dieticians with experience in providing care to metabolic patients.
Some children might need to be in special education classes, as intellectual disability is common with propionic academia.
Other treatment is symptomatic and supportive.
Genetic counseling is recommended for affected individuals and their families.
Information on current clinical trials is posted on the Internet at https://clinicaltrials.gov/ All studies receiving U.S. Government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Tollfree: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website: https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, contact: https://www.centerwatch.com/
For information about clinical trials conducted in Europe, contact: https://www.clinicaltrialsregister.eu/
1. Heringer J, et al. Impact of age at onset and newborn screening on outcome in organic acidurias. J Inherit Metab Dis. 2016; 39(3): 341-353.
2. Kolker S, et al, The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. J Inherit Metab Dis. 2015; 38(6): 1041-57.
3. Kolker S, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: the evolving clinical phenotype. J Inherit Metab Dis. 2015; 38(6):1059-74.
4. Fraser JL and Venditti CP. Methylmalonic and propionic acidemias: clinical management update. Curr Opin Pediatr. 2016; 28(6): 682-693.
5. Riemersma M, et al. Propionic acidemia as a cause of adult-onset dilated cardiomyopathy. Eur J Hum Genet. 2017; 25(11):1195-1201.
6. Tan, NS, et al. Metabolic cardiomyopathy from propionic acidemia precipitating cardiac arrest in a 25-year-old man. CMAJ. 2018; 190(29): E883-E887.
7. Pena L, et al. Natural history of propionic acidemia. Mol Genet Metab. 2012; 105(1): 5-9.
8. Grunert SC, et al. Propionic acidemia: clinical course and outcome in 55 pediatric and adolescent patients. Orphanet J Rare Dis. 2013; 8: 6.
9. Baumgartner MR, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis. 2014; 9:130.
10. Testai FD and Gorelick PB. Inherited metabolic disorders and stroke part 2: homocystinuria, organic acidurias, and urea cycle disorders. Arch Neurol. 2010; 67(2):148-53.
11. Lucke T, et al. Propionic acidemia: unusual course with late onset and fatal outcome. Metabolism. 2004; 53(6): 809-10
.
12. Wolf B, Paulsen EP and Hsia YE. Asymptomatic propionyl CoA carboxylase deficiency in a 13-year-old girl. J Pediatr. 1979; 95(4):563-5.
13. Sethi KD, et al. Adult-onset chorea and dementia with propionic acidemia. Neurology. 1989; 39(10): 343-5.
14. MacDonald A et al. Breast feeding in IMD. J Inherit Metab Dis. 2006; 29(2-3): 299-303.
15. Huner G, et al. Breastfeeding experience in inborn errors of metabolism other than phenylketonuria. J Inherit Metab Dis. 2005; 28(4):457-65.
16. Gallego-Villar L, et al. Functional characterization of novel genotypes and cellular oxidative stress studies in propionic acidemia. J Inherit Metab Dis. 2013; 36(5): 731-40.
17. Kraus JP, et al. Mutation analysis in 54 propionic acidemia patients. J Inherit Metab Dis. 2012; 35(1): 51-63.
18. Wongkittichote, P, Ah Mew N and Chapman KA. Propionyl-CoA carboxylase – A review. Mol Genet Metab. 2017; 122(4):145-152.
19. Wajner M and Goodman SI. Disruption of mitochondrial homeostasis in organic acidurias: insights from human and animal studies. J Bioenerg Biomembr. 2011; 43(1):31-8.
20. Rivera-Barahona A. et al. Treatment with antioxidants ameliorates oxidative damage in a mouse model of propionic acidemia. Mol Genet Metab. 2017; 122(1-2):43-50.
21. Shchelochkov OA, Carrillo N, Venditti C. Propionic Acidemia. 2012 May 17 [Updated 2016 Oct 6]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK92946/ Accessed November 1, 2018.
22. Couce ML, et al. Evaluation and long-term follow-up of infants with inborn errors of metabolism identified in an expanded screening programme. Mol Genet Metab. 2011; 104(4):470-5.
23. Schulze A. et al. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics. 2003; 111(6 Pt 1): 1399-406.
24. Chace DH, et al. Rapid diagnosis of methylmalonic and propionic acidemias: quantitative tandem mass spectrometric analysis of propionylcarnitine in filter-paper blood specimens obtained from newborns. Clin Chem. 2001; 47(11): 2040-4.
25. Dionisi-Vici C. et al. Inborn errors of metabolism in the Italian pediatric population: a national retrospective survey. J Pediatr. 2002; 140(3) 321-7.
26. Chapman KA, et al. Incidence of maple syrup urine disease, propionic acidemia, and methylmalonic aciduria from newborn screening data. Mol Genet Metab Rep. 2018;15: 106-109.
27. Al-Shamsi A, et al. Mutation Spectrum and Birth Prevalence of Inborn Errors of Metabolism among Emiratis: A study from Tawam Hospital Metabolic Center, United Arab Emirates. Sultan Qaboos Univ Med J. 2014; 14(1):e42-9.
28. Zayed H. Propionic acidemia in the Arab World Gene. 2015; 564(2):119-24.
29. Ravn K, et al. High incidence of propionic acidemia in greenland is due to a prevalent mutation, 1540insCCC, in the gene for the beta-subunit of propionyl CoA carboxylase. Am J Hum Genet. 2000; 67(1):203-6.
30. Grunert SC, et al. Propionic acidemia: neonatal versus selective metabolic screening. J Inherit Metab Dis. 2012; 35(1) 41-9.
31. Myles JG, Manoli I and Venditti CP. Effects of medical food leucine content in the management of methylmalonic and propionic acidemias. Curr Opin Clin Nutr Metab Care. 2018; 21(1):42-48.
32. Li M. et al. Cost-effectiveness of liver transplantation in methylmalonic and propionic acidemias. Liver Transpl. 2015; 21(9):1208-18.
33. Silva HM, et al. Liver Transplantation for Propionic Acidemia. J Pediatr Gastroenterol Nutr. 2017; 64(3):e73-e76.
34. Chapman KA, Summar ML, and Enns GM. Propionic acidemia: to liver transplant or not to liver transplant? Pediatr Transplant. 2012; 16(3): 209-10.

NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportOnline Mendelian Inheritance In Man (OMIM) has a summary of published research about this condition and includes references from the medical literature. The summary contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. OMIM is authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine.
View reportGeneReviews has an article on this condition covering diagnosis, management, and inheritance. Each article is written by one or more experts on the specific disease and is reviewed by other specialists. The article contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. The GeneReviews database is managed by the University of Washington.
View reportMedlinePlus has information about this condition that may include a description, frequency, causes, inheritance, and links to more information. The information is written for the public, including patients, caregivers and families. MedlinePlus is a service of the National Library of Medicine (NLM), which is part of the National Institutes of Health (NIH).
View reportPlease complete this form to access the requested resource.

