

Last updated:
01/18/2024
Years published: 1987, 1990, 1991, 1996, 1997, 1998, 1999, 2001, 2002, 2003, 2004, 2005, 2006, 2007, 2013, 2017, 2020
NORD gratefully acknowledges Arnold J. J. Reuser, PhD, emeritus professor in Cell Biology & Microscopical Anatomy, Erasmus University Medical Center, Rotterdam, the Netherlands, and John H. A. Dyck, adult patient, retired Professor in Political Science, Canada, for assistance in the preparation of this report.
Summary
Pompe disease is a rare disease continuum with variable rates of disease progression and different ages of onset. First symptoms can occur at any age from birth to late adulthood. Earlier onset compared to later onset is usually associated with faster progression and greater disease severity. At all ages, skeletal muscle weakness characterizes the disease, causing mobility problems and affecting the respiratory system.
The most severely affected infants usually present within the first 3 months after birth. They have characteristic heart (cardiac) problems (dysfunction due to heart enlargement) in addition to generalized skeletal muscle weakness and a life expectancy of less than 2 years, if untreated (classic infantile Pompe disease). Less severe forms of Pompe disease with onset during childhood, adolescence, or adulthood, rarely manifest cardiac problems, but gradually lead to walking disability and reduced respiratory function.
The scientific literature has different ways of subdividing the clinical spectrum of Pompe disease. Some articles describe ‘classic infantile’, ‘childhood’ and ‘adult’ Pompe disease while others discuss ‘infantile-onset’ (IOPD) and ‘late-onset’ (LOPD) disease.
Pompe disease is a rare, multisystemic, hereditary disease, which is caused by ‘pathogenic variations’ (abnormalities / mutations) in the ‘GAA gene’.
The GAA gene contains the genetic information for the production and function of a protein called ‘acid alpha-glucosidase’ (GAA). Shortage of this protein hampers the degradation of a complex sugar named ‘glycogen’ into a simple sugar named ‘glucose’. Therefore, glycogen starts to accumulate in all kinds of tissues, but primarily in skeletal muscle, smooth muscle and cardiac muscle, where it causes damage to tissue structure and function.
‘Enzyme replacement therapy’ (ERT), the only treatment presently available, aims to replenishing the shortage of GAA by intravenous administration of industrially made ‘rhGAA’ (recombinant human GAA).
Pompe disease is inherited in an autosomal recessive genetic pattern, which implies that healthy parents can have affected children.
Introduction
The human body can be seen as an assembly of interconnecting organs. Organs are composed of organ specific tissues, and tissues are composed of specialized cells like muscle cells, nerve cells, etc. Pompe disease belongs to a group of diseases known as the ‘lysosomal storage disorders’ (LSDs). Lysosomes are small compartments inside the cells wherein all kind of substances are re-cycled. The substances are degraded by the action of digestive enzymes. More than 50 different LSDs are presently known to be caused by the deficiency of one of these enzymes. Acid alpha-glucosidase (GAA) is one such enzyme and is responsible for the lysosomal degradation of glycogen. A shortage or dysfunction of GAA causes glycogen to accumulate within the lysosomes, which subsequently leads to cellular malfunction, cellular damage, tissue damage, and ultimately organ dysfunction. In Pompe disease, the organ dysfunction is mainly manifested by muscle weakness and muscle wasting.
Pompe disease is not only listed as an LSD, but also as one of the 15 presently known ‘glycogen storage disorders’ (GSDs), a group of metabolic disorders characterized by abnormalities in glycogen synthesis and breakdown.
Pompe disease is known under the alternative names ‘glycogen storage disease type II’ (GSDII), acid alpha-glucosidase (GAA) deficiency, and ‘acid maltase’ deficiency (acid maltase is another name for acid alpha-glucosidase).
Terminology
Pompe disease is divided into subtypes: ‘Classic infantile’ refers to the form of Pompe disease that was first described in 1932 and characterized by the onset of symptoms shortly after birth, generalized muscle weakness, and ‘cardiomegaly’ (a far too big heart), in combination with excessive glycogen stored in virtually all organs. Terms like ‘childhood’, ‘juvenile’, and ‘adult’ glycogenosis type II / Pompe disease / Acid maltase deficiency were historically introduced as names for the less severe forms of Pompe disease characterized by delayed onset and usually slower progression. Adult-onset was historically synonymous with ‘late-onset’.
After the introduction of enzyme replacement therapy, the meaning of ‘late-onset’ was increasingly referred to Pompe disease without hypertrophic cardiomyopathy (HCM) (thickened heart muscle).
Currently, the abbreviation IOPD (infantile-onset Pompe disease) refers in most but not all published cases to classic-infantile Pompe disease (some cases of childhood Pompe disease might be included). The abbreviation IPD (infantile Pompe disease) is also used. LOPD (late-onset Pompe disease) refers to all cases in which hypertrophic cardiomyopathy (HCM) did not manifest or was not diagnosed at or under the age of 1 year, as well as to all cases with symptom onset above the age of 1 year.
Patients with the ‘classic infantile’ form of Pompe disease are the most severely affected. Although hardly any symptoms may be apparent at birth, the disease usually presents within the first three months of life with rapidly progressive muscle weakness (‘floppy infants’), diminished muscle tone (hypotonia), respiratory deficiency, and a type of heart disease known as hypertrophic cardiomyopathy, a condition characterized by abnormal thickening of the walls of the heart (mainly the left chamber and the wall between the left and right chamber) resulting in diminished cardiac function. These problems together culminate in cardio-respiratory failure within the first 2 years of life.
Many infants have a large, protruding, tongue and a moderate enlargement of the liver. The legs often rest in a frog position and feel firm on palpation (pseudo-hypertrophy).
Feeding and swallowing problems as well as respiratory difficulties, which are often combined with respiratory tract infections, are common. Major developmental milestones such as rolling over, sitting up, and standing are delayed or not achieved. Mental development is usually normal. Virtually all infants experience hearing loss. The ‘classic infantile’ form of Pompe disease is caused by a total absence of acid alpha-glucosidase (GAA) activity and by a rapid buildup of glycogen in skeletal muscle and heart.
‘Childhood’ Pompe disease typically presents during childhood, and ‘adult’ Pompe disease during adulthood. In the current literature, these two forms of Pompe disease are often grouped together as ‘late-onset’ Pompe disease (abbreviated as LOPD) despite the fact that the time of presentation can vary from the first year of life to the eighth decade. Patients who develop symptoms early in life tend to be more severely affected and to have a faster rate of disease progression than those who develop symptoms later in life. Both children as well as adults usually have more GAA activity (their GAA deficiency is not total) than the most severely affected infants (who do not have any GAA activity), and the glycogen buildup is usually not as rapid. However, symptoms do progress, and can greatly affect the quality of life and diminish the lifespan.
Childhood and adult Pompe disease are associated with progressive weakness of mainly the proximal muscles (limb girdle, upper arms and upper legs), and varying degrees of respiratory weakness due to dysfunction of the diaphragm and the intercostal muscles (muscles between the ribs). The lower limbs are more affected than the upper limbs. The extent of muscle involvement is highly variable. Balance can be affected as the major leg muscles lose their strength and spring, forcing core muscles to take up the slack to maintain an upright posture. The muscles adjacent to the spinal column (para-spinal muscles) and neck are usually also involved. Weakness of the para-spinal muscles around puberty can cause abnormal curvature of the spine (scoliosis). Due to the combination of these serious symptoms, affected individuals may become wheelchair and/or ventilator dependent.
Other symptoms can include chewing and swallowing difficulties and drooping of the upper eyelids (ptosis). Additionally, blood vessel abnormalities due to smooth muscle weakness and problems of the urinary and digestive systems have been reported.
Pompe disease is inherited in an autosomal recessive pattern. Recessive genetic disorders occur when an individual inherits a non-working gene from each parent. If an individual receives one working gene and one non-working gene for the disease, the person will be a carrier for the disease. Pompe disease carriers will not show symptoms. The risk for two carrier parents to both pass the non-working gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier, like the parents, is 50% with each pregnancy. The chance for a child to receive working genes from both parents is 25%. The risk is the same for males and females.
Pompe disease is caused by pathogenic variations (mutations) in the acid alpha-glucosidase (GAA) gene. Close to 600 different GAA gene variations have been identified in families with this disorder. All the known variations of the GAA gene are collected and listed in the Pompe variant database at: www.pompevariantdatabase.nl along with a description of how detrimental they are and with which clinical forms of Pompe disease they are associated.
The degree of acid alpha-glucosidase (GAA) deficiency is dictated by the nature of the variations in each of the 2 GAA gene copies (1 from the father with variation A and 1 from the mother with variation B) and their combined effect. Generally: the more GAA deficiency these variants are causing, the earlier the onset of symptoms, the faster the disease progression, and the greater the clinical severity. However, the clinical presentation of Pompe disease is not solely dictated by the nature of the inherited pathogenic variations in the 2 GAA gene copies, but additionally influenced by a number of still unknown genetic, epigenetic, and environmental factors. These latter might include diet, lifestyle, exercise, etc.
Pompe disease occurs in various populations and ethnic groups around the world. Estimates vary, but its incidence is generally placed at approximately 1 in 40,000 births in the United States (and in the Netherlands). However, a recent review of birth incidences in Missouri reported a much higher incidence of 1 in 5,463 in that state. A ‘founder effect’ cannot be excluded. The same holds for Pompe disease in the American Black population and for Pompe disease in Taiwan where unique pathogenic variants have been reported.
Most physicians are not familiar with Pompe disease. They may never have had patients with Pompe Disease. They need to know what they are looking for. The diagnosis of Pompe disease is based on a thorough clinical evaluation, a detailed patient and family history, and a variety of biochemical tests with first of all the measuring of GAA activity. Preimplantation testing and prenatal diagnosis are also possible when a pregnancy is known to be at risk for Pompe disease.
Clinical Testing and Work Up
In individuals suspected of having Pompe disease, blood can be drawn and the function/activity of GAA (the ‘enzymatic activity’) can be measured in white blood cells (leukocytes), but only if the proper assay conditions are being used and acarbose is added to the reaction mixture to inhibit the activity of glucoamylase. The isolation of lymphocytes to prevent the interference of glucoamylase is not advised, as the successful isolation of lymphocytes is not only time consuming, but also error prone when the blood sample is not sufficiently fresh.
Alternatively, the GAA activity/functional assay can also be performed on dried blood spots, but this method is not any quicker, less reliable, and also requires the use of acarbose to inhibit the glucoamylase activity. An important feature of the bloodspot test method is that it allows convenient shipment of samples if a certified diagnostic laboratory test is not locally available. Moreover, the dried blood spot test is unquestionably the most convenient methodology for the screening of large populations of newborns and large numbers of patients with undiagnosed limb-girdle muscular dystrophies and unexplained ‘CK-emias’ (a high level of creatine kinase in the blood pointing to muscle damage).
Each diagnosis performed with the dried blood spot test method must be confirmed through molecular genetic testing (GAA gene copy analysis) or by measuring the GAA activity with another method. Leukocytes can be used for this purpose, but cultured skin fibroblasts obtained by a skin biopsy are the very best material. More invasive muscle biopsies are not needed and not optimal either for measuring the GAA activity.
The advantage of measuring the GAA activity/function is that the finding of a clear GAA deficiency testifies of Pompe disease even if the pathogenic variations in the 2 GAA gene copies are not found.
The advantage of searching for pathogenic variations in the 2 GAA gene copies (a method called ‘DNA analysis’) is that only this test discriminates unequivocally between carriers and affected children/adults within families (measuring the GAA activity/function may not do so). It is advised to use both test methods if affordable and technically possible.
The application of a skin biopsy and the initiation of a culture of skin fibroblasts might not be feasible in every diagnostic setting, but should always be considered as there are important advantages with this procedure. The GAA activity/functional test using skin fibroblasts is superior to all other methods for its high sensitivity and for discriminating between classic-infantile, childhood and adult Pompe disease (IOPD vs LOPD) in almost all cases. Cultured skin fibroblasts can be stored forever and be used as eternal sample source for measuring GAA activity, searching for GAA variations, studying defects in GAA production, and for the development of therapeutic interventions.
A variety of other tests can be performed to detect or assess symptoms potentially associated with Pompe disease such as sleep studies, tests that measure lung function, and tests that measure muscle function. Muscle MRI (imaging by magnetic resonance) is used to visualize the degree of muscle damage.
Specific tests may also be performed to assess the heart function, including chest x-ray, electrocardiography (ECG), and echocardiography (imaging by ultrasound). Chest x-rays allow physicians to assess the size of the heart, which is enlarged in classic infantile Pompe disease. Electrocardiography (ECG) measures the electric activity of the heart and detects abnormal heart rhythms. Echocardiography uses reflected sound waves to create a picture of the heart and can reveal abnormal thickening of the walls of the heart.
When the pathogenic GAA variations of both parents are known, pre-implantation diagnosis and prenatal diagnosis are possible. The latter is preferably performed by chorionic villi sampling (CVS) in early pregnancy. Pre-implantation genetic diagnosis (PGD: testing in a very early embryonic stage to determine whether the embryo has inherited the pathogenic variations from the parents) may also be an option. PGD is performed on embryos created through in vitro fertilization (IVF). Families interested in PGD should seek the counsel of a certified genetic counselor.
Not all individuals with Pompe disease are timely diagnosed and too many are, unfortunately, misdiagnosed. Since the introduction of the dried blood spot test method, formerly undiagnosed patients were identified in screening programs among individuals with limb-girdle dystrophies and/or ‘hyper CK-emia’ of unknown cause (a high level of creatine kinase in the blood is indicative of muscle damage).
Newborn Screening
The introduction of newborn screening (NBS) in some US states and countries, using dried blood spots for first tier testing, has contributed significantly to the early detection and treatment of Pompe disease patients. But, NBS has the draw back that all clinical forms of Pompe disease are detected at birth while only the most severely affected patients with classic-infantile Pompe disease / IOPD will develop immediate symptoms and are likely to succumb before the age of 1 year, if untreated. Families in which newborns are diagnosed with LOPD, encompassing childhood and adult forms, remain uncertain about their child’s future since symptoms in the newborn may manifest around the age of 1 year or several decades later.
The link between GAA variations and clinical phenotypes emerging from the systematic collection of GAA variations and clinical forms in databases such as the Pompevariant database www.pompevariantdatabase.nl and the Pompe Registry is expected to provide future guidance.
Treatment
The treatment of Pompe disease is disease-specific, symptomatic and supportive. Treatment requires the coordinated efforts of a team of specialists. The input of pediatricians, internists, neurologists, orthopedists, cardiologists, dieticians, physical therapists and other healthcare professionals may be needed to develop the treatment plan. The treatment plan must be patient centered, including the patient’s self-descriptions or caregiver’s descriptions (patient history), which provide important data for the team of specialists. A patient or caregiver who is fully informed can provide better experiential data. Genetic counseling is of crucial importance for affected individuals and their families.
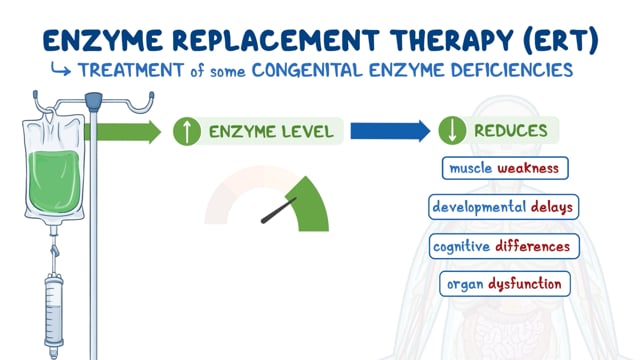
Enzyme Replacement Therapy
Enzyme replacement therapy (ERT) is an approved treatment for all patients with Pompe disease. It involves the intravenous administration of recombinant human acid alpha-glucosidase (rhGAA). This treatment is called Lumizyme (marketed as Myozyme outside the United States) and was first approved by the U.S. Food and Drug Administration (FDA) in 2006. ERT has been shown to extend the life expectancy of patients with infantile-onset Pompe disease, but these patients are not fully cured and residual symptoms remain. There is some evidence that the currently prescribed and approved dosage is not sufficient in all patients.
Most patients with childhood and adult forms of Pompe disease also benefit from ERT. In 2021, avalglucosidase alfa-ngpt (Nexviazyme) was approved by the FDA to treat patients one year of age and older with late-onset Pompe disease. This enzyme replacement therapy is an intravenous medication that helps reduce the accumulation of glycogen.
In 2023, the combination therapy of cipaglucosidase alfa-atga (Pombiliti) and miglustat (Opfolda) was FDA approved to treat adults with late-onset Pompe disease who weigh ≥40 kg and are not improving on their current enzyme replacement therapy.
Supportive Therapies
Additional treatment of Pompe disease is symptomatic and supportive. Respiratory support may be required, as most patients have some degree of respiratory compromise and/or respiratory failure. Physical therapy may be helpful to strengthen respiratory muscles. Some patients may need respiratory assistance through mechanical ventilation (i.e. Bipap or volume ventilators) during the night and/or periods of the day or during respiratory tract infections. Mechanical ventilation support can be through noninvasive or invasive techniques. Decisions about the duration of respiratory support are best made by the patients themselves or the parents in careful consultation with the patient’s physicians and other members of the healthcare team. Physiotherapy is recommended to improve strength and physical ability. Occupational therapy, including the use of canes or walkers, may be necessary. Eventually, some patients may require the use of a wheelchair. Speech therapy can be beneficial in some patients to improve articulation and speech. Orthopedic devices including braces may be recommended in some patients. Surgery may be required for certain orthopedic symptoms such as contractures or spinal deformity.
Since Pompe disease can weaken muscles used for chewing and swallowing, adequate measures may be required to ensure proper nutrition and weight gain. Some patients may need specialized, high-calorie diets and may need to learn techniques to change the size and texture of food to lower the risk of aspiration. Some infants may require the insertion of a feeding tube that is run through the nose, down the esophagus and into the stomach (nasogastric tube). In some children, a feeding tube may need to be inserted directly into the stomach through a small surgical opening in the abdominal wall. Some individuals with childhood or adult Pompe disease / LOPD may require a soft diet, but few require feeding tubes.
Gene therapy remains an exciting option and progress continues in its development. Gene therapy in Pompe disease is directed toward restoring the acid alpha-glucosidase (GAA) production and activity in crucial tissues like the diaphragm to improve respiratory capacity. Other gene therapy efforts seek to restore the body’s ability to produce acid alpha-glucosidase (GAA) by transducing a ‘normal’ GAA gene copy in the patient’s liver cells (via intravenous administration) or in bone marrow stem cells -taken out of the body, modified outside the body, and put back into the body- (HSCGT; hematopoietic stem cell gene therapy). At present, multiple groups are working to advance gene therapy to the clinical trial stage.
Improvements to the industrial rhGAA currently used for ERT are being explored. For example, carbohydrate side chains of rhGAA are modified to improve uptake by muscle cells, and drugs (chaperones) are employed to stabilize the intravenously administered form of rhGAA.
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov . All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Toll-free: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website:
https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, in the main, contact:
www.centerwatch.com
For information about clinical trials conducted in Europe, contact:
https://www.clinicaltrialsregister.eu/
JOURNAL ARTICLES
Clinical
van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, Smit GP, Poll-The BT, Bakker HD, Loonen MC, de Klerk JB, Reuser AJ, van der Ploeg AT..The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332-340. PMID: 12897283
doi: 10.1542/peds.112.2.332
Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671-676. PMID: 16737883
doi: 10.1016/j.jpeds.2005.11.033
Carine I van Capelle 1, Andre Goedegebure, Nienke C Homans, Hans L J Hoeve, Arnold J Reuser, Ans T van der Ploeg Hearing loss in Pompe disease revisited: results from a study of 24 children. J Inherit Metab Dis. 2010;33:597-602. PMID: 20596893 PMCID: PMC2946566
doi: 10.1007/s10545-010-9144-0
Winkel LP, Hagemans ML, van Doorn PA, Loonen MC, Hop WJ, Reuser AJ, van der Ploeg AT. The natural course of non-classic Pompe’s disease; a review of 225 published cases. J Neurol. 2005 Aug;252(8):875-884. PMID: 16133732
doi: 10.1007/s00415-005-0922-9
Hagemans ML, Winkel LP, Hop WC, Reuser AJ, Van Doorn PA, Van der Ploeg AT. Disease severity in children and adults with Pompe disease related to age and disease duration. Neurology. 2005;64:2139-2141. PMID: 15985590
doi: 10.1212/01.WNL.0000165979.46537.56
Deniz Güngör, Juna M de Vries, Wim C J Hop, Arnold J J Reuser, Pieter A van Doorn, Ans T van der Ploeg, Marloes L C Hagemans. Survival and associated factors in 268 adults with Pompe disease prior to treatment with enzyme replacement therapy. Orphanet J Rare Dis. 2011;6:34. PMID: 21631931 PMCID: PMC3135500
doi: 10.1186/1750-1172-6-34
Nadine A M E van der Beek, Juna M de Vries, Marloes L C Hagemans, Wim C J Hop, Marian A Kroos, John H J Wokke, Marianne de Visser, Baziel G M van Engelen, Jan B M Kuks, Anneke J van der Kooi, Nicolette C Notermans, Karin G Faber, Jan J G M Verschuuren, Arnold J J Reuser, Ans T van der Ploeg, Pieter A van Doorn. Clinical features and predictors for disease natural progression in adults with Pompe disease: a nationwide prospective observational study.Orphanet J Rare Dis. 2012;7:88. PMID: 23147228 PMCID: PMC3551719
doi: 10.1186/1750-1172-7-88
Incidence + Founder Effect
Ausems MG, Verbiest J, Hermans MP, Kroos MA, Beemer FA, Wokke JH, Sandkuijl LA, Reuser AJ, van der Ploeg. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counseling. Eur J Hum Genet. 1999;7:713-716. PMID: 10482961
doi: 10.1038/sj.ejhg.5200367
Huie ML, Chen AS, Tsujino S, Shanske S, DiMauro S, Engel AG, Hirschhorn R. Aberrant splicing in adult onset glycogen storage disease type II (GSDII): molecular identification of an IVS1 (-13T–>G) mutation in a majority of patients and a novel IVS10 (+1GT–>CT) mutation. Hum Mol Genet. 1994;3:2231-2236. PMID: 7881425
doi: 10.1093/hmg/3.12.2231
Ausems MG, ten Berg K, Sandkuijl LA, Kroos MA, Bardoel AF, Roumelioti KN, Reuser AJ, Sinke R, Wijmenga C. Dutch patients with glycogen storage disease type II show common ancestry for the 525delT and del exon 18 mutations. J Med Genet. 2001;38:527-529. PMID: 11494962 PMCID: PMC1734921
doi: 10.1136/jmg.38.8.527
Becker JA, Vlach J, Raben N, Nagaraju K, Adams EM, Hermans MM, Reuser AJ, Brooks SS, Tifft CJ, Hirschhorn R, Huie ML, Nicolino M, Plotz PH. The African origin of the common mutation in African American patients with glycogen-storage disease type II. Am J Hum Genet. 1998;62:991-994. PMID: 9529346 PMCID: PMC1377028
doi: 10.1086/301788
Shieh JJ, Lin CY. Frequent mutation in Chinese patients with infantile type of GSD II in Taiwan: evidence for a founder effect. Hum Mutat. 1998;11:306-312. PMID: 9554747
doi: 10.1002/(SICI)1098-1004(1998)11:4<306::AID-HUMU8>3.0.CO;2-S
Patricia L. Hall,, Rossana Sanchez, Arthur F. Hagar, S. Caleb Jerris, Angela Wittenauer, and William R. Wilcox.Two-tiered Newborn Screening with post-analytical tools for Pompe disease and Mucopolysaccharidosis Type I. Results in Performance Improvement and Future Direction. Int. J. Neonatal Screen. 2020, 6(1). PMID: 32064362 PMCID: PMC7021244
doi:10.3390/ijns6010002
Diagnosis
Pompe Disease Diagnostic Working Group; B Winchester, D Bali, O A Bodamer, C Caillaud, E Christensen, A Cooper, E Cupler, M Deschauer, K Fumić, M Jackson, P Kishnani, L Lacerda, J Ledvinová, A Lugowska, Z Lukacs, I Maire, H Mandel, E Mengel, W Müller-Felber, M Piraud, A Reuser, T Rupar, I Sinigerska, M Szlago, F Verheijen, O P van Diggelen, B Wuyts, E Zakharova, J Keutzer. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus meeting. Mol Genet Metab. 2008;93:275-281. PMID: 18078773 Epub 2007 Dec 19.
doi: 10.1016/j.ymgme.2007.09.006
Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, Crowley JF, Downs S, Howell RR, Kravitz RM, Mackey J, Marsden D, Martins AM, Millington DS, Nicolino M, O’Grady G, Patterson MC, Rapoport DM, Slonim A, Spencer CT, Tifft CJ, Watson MS. Pompe disease diagnosis and management guideline. Genet Med. 2006;8:267-288. PMID: 16702877 PMC3110959
doi: 10.1097/01.gim.0000218152.87434.f3
Niño MY, Wijgerde M, de Faria DOS, Hoogeveen-Westerveld M, Bergsma AJ, Broeders M, van der Beek NAME, van den Hout HJM, van der Ploeg AT, Verheijen FW, Pijnappel WWMP. Enzymatic diagnosis of Pompe disease: lessons from 28 years of experience. Eur J Hum Genet. 2020. Nov 8. Online ahead of print.
PMID: 33162552 doi: 10.1038/s41431-020-00752-2
Chafic Karam, Thomas Ragole, Orly Moshe-Lilie, Nizar Chahin. Unwarranted, long term, Alglucosidase Alfa enzyme replacement therapy in two non-Pompe disease patients. Clin Neurol Neurosurg. 2020;196:106048. PMID: 32623214
doi: 10.1016/j.clineuro.2020.106048
ERT
H Van den Hout, A J Reuser, A G Vulto, M C Loonen, A Cromme-Dijkhuis, A T Van der Ploeg. Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet 2000;356:397-398. PMID: 10972374
doi: 10.1016/s0140-6736(00)02533-2
Kishnani PS, Nicolino M, Voit T, Rogers RC, Tsai AC, Waterson J, Herman GE, Amalfitano A,Thurberg BL, Richards S, Davison M, Corzo D, Chen YT. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006;149:89–97. PMID: 16860134 PMCID: PMC2692727
doi: 10.1016/j.jpeds.2006.02.03
Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68(2):99-109. PMID: 17151339 Epub 2006 Dec 6
doi: 10.1212/01.wnl.0000251268.41188.04
van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ, Herson S, Kishnani PS, Laforet P, Lake SL, Lange DJ, Leshner RT, Mayhew JE, Morgan C, Nozaki K, Park DJ, Pestronk A, Rosenbloom B, Skrinar A, van Capelle CI, van der Beek NA, Wasserstein M, Zivkovic SA. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med. 2010;362:1396-1406. PMID: 20393176
doi: 10.1056/NEJMoa0909859
Long-term follow up of infants
van Capelle CI, Poelman E, Frohn-Mulder IM, Koopman LP, van den Hout JMP, Régal L, Cools B, Helbing WA, van der Ploeg AT. Cardiac outcome in classic infantile Pompe disease after 13 years of treatment with recombinant human acid alpha-glucosidase. Int J Cardiol. 2018;269:104-110. PMID: 30049495 Epub 2018 Jul 19
doi: 10.1016/j.ijcard.2018.07.091
Spiridigliozzi GA, Keeling LA, Stefanescu M, Li C, Austin S, Kishnani PS. Cognitive and academic outcomes in long-term survivors of infantile-onset Pompe disease: A longitudinal follow-up. Mol Genet Metab. 2017;121:127-137. PMID: 28495044 Epub 2017 May 1
doi: 10.1016/j.ymgme.2017.04.014
Ebbink BJ, Poelman E, Aarsen FK, Plug I, Régal L, Muentjes C, van der Beek NAME, Lequin MH, van der Ploeg AT, van den Hout JMP. Classic infantile Pompe patients approaching adulthood: a cohort study on consequences for the brain. Dev Med Child Neurol. 2018;60:579-586. PMID: 29573408 Epub 2018 Mar 24.
doi: 10.1111/dmcn.13740
Parini R, De Lorenzo P, Dardis A, et al. Long term clinical history of an Italian cohort of infantile onset Pompe disease treated with enzyme replacement therapy. Orphanet J Rare Dis. 2018;13:32. PMID: 29422078 PMCID: PMC5806382
doi: 10.1186/s13023-018-0771-0
Gupta N, Kazi ZB, Nampoothiri S, et al. Clinical and molecular disease spectrum and outcomes in patients with infantile-onset Pompe Disease. Pediatr. 2020;216:44-50.e5. PMID: 31606152 Epub 2019 Oct 9
doi: 10.1016/j.jpeds.2019.08.058
Hahn A, Schänzer A. Long-term outcome and unmet needs in infantile-onset Pompe disease. Ann Transl Med. 2019;7(13):283. PMID: 31392195 PMCID: PMC6642934
doi: 10.21037/atm.2019.04.70
Poelman E, van den Dorpel JJA, Hoogeveen-Westerveld M, van den Hout JMP, van der Giessen LJ, van der Beek NAME, Pijnappel WWMP, van der Ploeg AT. Effects of higher and more frequent dosing of alglucosidase alfa and immunomodulation on long-term clinical outcome of classic infantile Pompe patients. J Inherit Metab Dis. 2020 Jun 7. PMID: 32506446
doi: 10.1002/jimd.12268 Online ahead of print.
Khan AA, Case LE, Herbert M, et al. Higher dosing of alglucosidase alfa improves outcomes in children with Pompe disease: a clinical study and review of the literature. Genet Med. 2020;22:898-907. PMID: 31904026 Epub 2020 Jan 6.
doi: 10.1038/s41436-019-0738-0
Kazi ZB, Desai AK, Berrier KL, et al. Sustained immune tolerance induction in enzyme replacement therapy-treated CRIM-negative patients with infantile Pompe disease. JCI Insight. 2017;2(16):e94328. PMID: 28814660 PMCID: PMC5621909
doi: 10.1172/jci.insight.94328
Poelman E, Hoogeveen-Westerveld M, Kroos-de Haan MA, van den Hout JMP, Bronsema KJ, van de Merbel NC, van der Ploeg AT, Pijnappel WWMP. High sustained antibody titers in patients with classic infantile Pompe disease following immunomodulation at start of enzyme replacement therapy. J Pediatr. 2018;195:236-243e233. PMID: 29428273 Epub 2018 Feb 7
doi: 10.1016/j.jpeds.2017.11.046
Long-term follow up of children and adults
van der Meijden JC, Kruijshaar ME, Harlaar L, Rizopoulos D, van der Beek NAME, van der Ploeg AT. Long-term follow-up of 17 patients with childhood Pompe disease treated with enzyme replacement therapy. J Inherit Metab Dis. 2018;41:1205-1214. PMID: 29556838 PMCID PMC6326992
doi: 10.1007/s10545-018-0166-3
Hahn SH, Kronn D, Leslie ND, Pena LDM, Tanpaiboon P, Gambello MJ, Gibson JB, Hillman R, Stockton DW, Day JW, Wang RY, An Haack K, Shafi R, Sparks S, Zhao Y, Wilson C, Kishnani PS; Pompe ADVANCE Study Consortium Efficacy, safety profile, and immunogenicity of alglucosidase alfa produced at the 4,000-liter scale in US children and adolescents with Pompe disease: ADVANCE, a phase IV, open-label, prospective study. Genet Med. 2018;20:1284-1294. PMID: 29565424 Epub 2018 Mar 22.
doi: 10.1038/gim.2018.2
Harlaar L, Hogrel J-Y, Perniconi B, et al. Large variation in effects during 10 years of enzyme therapy in adults with Pompe disease. Neurology. 2019;93(19):e1756-e1767. PMID: 31619483 PMCID: PMC6946483
doi: 10.1212/WNL.0000000000008441 Epub 2019 Oct 16.
van der Ploeg AT, Kruijshaar ME, Toscano A, et al. European Pompe Consortium. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience. Eur J Neurol. 2017;24:768-e31. PMID: 28477382 doi: 10.1111/ene.13285 Epub 2017 May 6.
NBS
Li Y, Scott CR, Chamoles NA, Ghavami A, Pinto BM, Turecek F, Gelb MH. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem. 2004;50:1785-96. PMID: 15292070 PMCID: PMC3428798
doi: 10.1373/clinchem.2004.035907 Epub 2004 Aug 3
Kishnani PS, Hwu W-L and on behalf of the Pompe Disease Newborn Screening Working Group. Introduction to the Newborn Screening, diagnosis, and treatment for Pompe disease Guidance Supplement. Pediatrics July 2017, 140 (Supplement 1) S1-S3.
Doi: 10.1542/peds.2016-0280B
Hopkins PV, Campbell C, Klug T, Rogers S, Raburn-Miller J, Kiesling J. Lysosomal storage disorder screening implementation: findings from the first six months of full population pilot testing in Missouri. J Pediatr. 2015 Jan;166:172-177. PMID: 25444528 Epub 2014 Oct 18.
doi: 10.1016/j.jpeds.2014.09.023
Chien W-H, Hwu W-L, Lee N-C. Newborn screening: Taiwanese experience. Ann Transl Med. 2019;7(13):281. PMID: 31392193 PMCID: PMC6642927
doi: 10.21037/atm.2019.05.47
Hall PL, Sanchez R, Hagar AF, Jerris SC, Wittenauer A, and Wilcox WR. Two-tiered Newborn Screening with post-analytical tools for Pompe disease and Mucopolysaccharidosis Type I. Results in performance improvement and future direction. Int. J. Neonatal Screen. 2020, 6(1). PMID: 32064362 PMCID: PMC7021244
doi:10.3390/ijns6010002
Atherton AM, Day-Salvatore D. Pompe Disease Newborn Screening Working Group. The role of Genetic Counseling in Pompe disease after patients are identified through Newborn Screening. Pediatrics. 2017;140(Suppl 1):S46-S50. PMID: 29162676
doi: 10.1542/peds.2016-0280F
Kronn DF, Day-Salvatore D, Hwu WL, Jones SA, Nakamura K, Okuyama T, Swoboda KJ, Kishnani PS; Pompe Disease Newborn Screening Working Group. Management of confirmed Newborn-Screened patients with Pompe disease across the disease spectrum. Pediatrics. 2017;140(Suppl 1):S24-S45. PMID: 29162675
doi: 10.1542/peds.2016-0280E
Pruniski & E. Lisi & N. Ali. Newborn screening for Pompe disease: impact on families. J Inherit Metab Dis. 2018;41:1189-1203. PMID: 29594646 Epub 2018 Mar 28.
doi: 10.1007/s10545-018-0159-2.
Bombard Y, Miller FA, Hayeems RZ, Avard D, Knoppers BM, Cornel MC, Borry P. The expansion of newborn screening: is reproductive benefit an appropriate pursuit? Nat Rev Genet. 2009;10:666-667. PMID: 19763150
doi: 10.1038/nrg2666
Counseling
Ausems MG, ten Berg K, Beemer FA and Wokke JH. Phenotypic expression of late-onset glycogen storage disease Type II: identification of asymptomatic adults through family studies and review of reported families. Neuromuscul Disord. 2000;10:467-471. PMID: 10996774
doi: 10.1016/s0960-8966(00)00123-1
Wens SCA, van Gelder CM, Kruijshaar MR de Vries JM, van der Beek NAME, Reuser AJJ, van Doorn PA, van der Ploeg AT, Brusse E. Phenotypical variation within 22 families with Pompe disease. Orphanet J Rare Dis. 2013 Nov 19;8:182. PMID: 24245577 PMCID: PMC3843594
doi: 10.1186/1750-1172-8-182
Atherton AM, Day-Salvatore D. Pompe Disease Newborn Screening Working Group. The role of Genetic Counseling in Pompe disease after patients are identified through Newborn Screening. Pediatrics. 2017;140(Suppl 1):S46-S50. PMID: 29162676
doi: 10.1542/peds.2016-0280F
Databases
Niño MY, In’t Groen SLM Bergsma AJ, van der Beek NAME, Kroos M, Hoogeveen-Westerveid M, van der Ploeg AT, Pijnappel WWMP. Extension of the Pompe mutation database by linking disease-associated variants to clinical severity. Hum Mutat. 2019;40:1954-1967. PMID: 31254424 PMCID: PMC6851659 doi: 10.1002/humu.23854 Epub 2019 Jul 29.
Reuser AJJ, van der Ploeg AT, Chien Y-S, et al. On Behalf Of The Pompe Registry Sites. GAA variants and phenotypes among 1,079 patients with Pompe disease: Data from the Pompe Registry.Hum Mutat. 2019;40:2146-2164. PMID: 31342611 PMCID: PMC6852536
doi: 10.1002/humu.23878 Epub 2019 Aug 7
The Pompe variant database at: https://www.pompevariantdatabase.nl/pompe_mutations_list.php?orderby=aMut_ID1
Gene therapy
Corti M, Liberati C, Smith BK, et al. Safety of Intradiaphragmatic Delivery of Adeno-Associated Virus-Mediated Alpha-Glucosidase (rAAV1-CMV-hGAA) Gene Therapy in Children Affected by Pompe Disease. Hum Gene Ther Clin Dev. 2017;28:208-218. PMID: 29160099 PMCID: PMC5733674
doi: 10.1089/humc.2017.146
Cagin U, Puzzo F, Gomez MJ, Moya-Nilges M, Sellier P, Abad C, Van Wittenberghe L, Daniele N, Guerchet N, Gjata B, Collaud F, Charles S, Sola MS, Boyer O, Krijnse- Locker J, Ronzitti G, Colella P, Mingozzi. Rescue of advanced Pompe disease in mice with hepatic expression of secretable acid α-glucosidase. Molecular Therapy 2020 May 30;S1525-0016(20)30289-6 PMID: 32526204
doi: https://doi.org/10.1016/ j.ymthe.2020.05.025
Lim J-A, Yi H, Gao F, Raben N, Kishnani PS, Sun B. Intravenous injection of an AAV-PHP.B vector encoding human acid α-Glucosidase rescues both muscle and CNS defects in Murine Pompe disease.Mol Ther Methods Clin Dev. 2019;12:233-245. PMID: 30809555 PMCID: PMC6376130
doi: 10.1016/j.omtm.2019.01.006 eCollection 2019 Mar 15.
Stok M, de Boer H, Huston MW, Jacobs EH, Roovers O, Visser P, Jahr H, Duncker DJ, van Deel ED, Reuser AJJ, van Til NP, Wagemaker G. Lentiviral Hematopoietic Stem Cell Gene Therapy corrects Murine Pompe disease.Mol Ther Methods Clin Dev. 2020;17:1014-1025. eCollection 2020 Jun 12PMID: 32462050 PMCID: PMC7240064
doi: 10.1016/j.omtm.2020.04.023
Piras G, Montiel-Equihua C, Chan YA, Wantuch S, Stuckey D, Burke D, Prunty H, Phadke R, Chambers D, Partida-Gaytan A, Leon-Rico D, Panchal N, Whitmore K, Calero M, Benedetti S, Santilli G, Thrasher AJ, Gaspar HB. Lentiviral Hematopoietic Stem Cell Gene Therapy rescues clinical phenotypes in a Murine model of Pompe disease. Mol Ther Methods Clin Dev. 2020;18:558-570. PMID: 32775491 PMCID: PMC7396971
doi: 10.1016/j.omtm.2020.07.001 eCollection 2020 Sep 11.
van der Wal E, Bergsma AJ, Pijnenburg JM, van der Ploeg AT, Pijnappel WWMP. Antisense oligonucleotides promote exon inclusion and correct the common c.-32-13T>G GAA splicing variant in Pompe disease. Mol Ther Nucleic Acids. 2017;7:90-100. PMID: 28624228 PMCID: PMC5415969
doi: 10.1016/j.omtn.2017.03.001 Epub 2017 Mar 14.
Goina E, Peruzzo P, Bembi B, Dardis A, Buratti E.Glycogen reduction in myotubes of late-onset Pompe disease patients using Antisense Technology. Mol Ther. 2017;25:2117-2128. PMID: 28629821 PMCID: PMC5589062
doi: 10.1016/j.ymthe.2017.05.019 Epub 2017 Jun 16
INTERNET
BOOK CHAPTER
Arnold J. J. Reuser, Rochelle Hirschhorn, Marian A. Kroos
Pompe disease: Glycogen Storage Disease Type II, Acid α-Glucosidase (Acid Maltase) Deficiency; 65 pages, 623 references
The Online Metabolic and Molecular Bases of Inherited Disease (2018)
Part 16, Chapter 135 doi: 10.1036/ommbid.417

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportOnline Mendelian Inheritance In Man (OMIM) has a summary of published research about this condition and includes references from the medical literature. The summary contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. OMIM is authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine.
View reportGeneReviews has an article on this condition covering diagnosis, management, and inheritance. Each article is written by one or more experts on the specific disease and is reviewed by other specialists. The article contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. The GeneReviews database is managed by the University of Washington.
View reportMedlinePlus has information about this condition that may include a description, frequency, causes, inheritance, and links to more information. The information is written for the public, including patients, caregivers and families. MedlinePlus is a service of the National Library of Medicine (NLM), which is part of the National Institutes of Health (NIH).
View reportPlease complete this form to access the requested resource.
