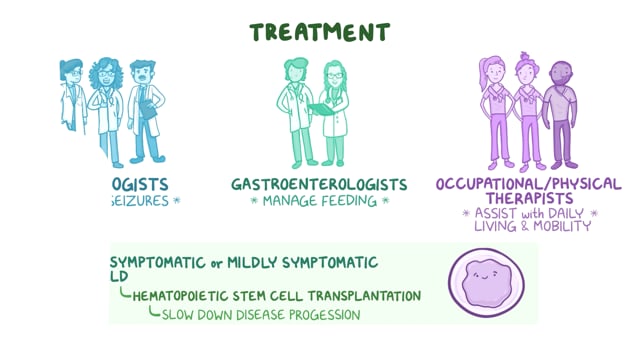
Tay-Sachs disease is a rare, neurodegenerative disorder in which deficiency of an enzyme (b-hexosaminidase A) results in excessive accumulation of certain fats (lipids) known as gangliosides in the brain tissue and certain organs. This abnormal accumulation of gangliosides leads to progressive dysfunction of the central nervous system. Tay-Sachs disease is categorized as a lysosomal storage disease. Lysosomes are the major digestive units in cells. Enzymes within lysosomes break down or “digest” complex molecules such as mixtures of carbohydrates and fats (like glycosphingolipids). When one of these lysosomal enzymes (such as b-hexosaminidase A) is missing or ineffective, glycosphingolipids start to build up in the lysosome. If there is too much accumulation of these materials in the lysosome, the cells in the nervous system degenerate and die, triggering an inflammatory response that amplifies damage in surrounding tissue.
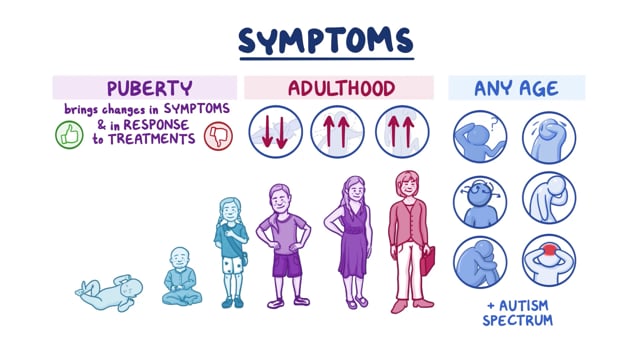
The most common form of Tay-Sachs disease is the Infantile form, which can present around 6 months of age as reduced vision and an exaggerated startle response and eventually progress to a gradual loss of acquired skills and seizures by age 2 and leads early death, usually by the age of 5. There is also a juvenile version of the disease beginning at about the age of 5 years of age and adult forms of Tay-Sachs disease also known as late-onset Tay Sachs disease (LOTS) beginning in the late teens and beyond. All three forms of Tay-Sachs disease are inherited in an autosomal recessive manner and the age of onset is a function of the amount, if any, of residual enzyme activity.
Please complete this form to access the requested resource.