

Last updated:
1/27/2025
Years published: 1984, 1985, 1986, 1987, 1988, 1989, 1992, 1997, 1998, 1999, 2000, 2001, 2002, 2017, 2021, 2025
NORD gratefully acknowledges Kathleen Renna, BS, and Camilo Toro, MD, NIH Undiagnosed Diseases Program, National Human Genome Research Institute, National Institutes of Health, for assistance in the preparation of this report.
Tay-Sachs disease is a rare, neurodegenerative disorder in which deficiency of an enzyme (b-hexosaminidase A) results in excessive accumulation of certain fats (lipids) known as gangliosides in the brain tissue and certain organs. This abnormal accumulation of gangliosides leads to progressive dysfunction of the central nervous system. Tay-Sachs disease is categorized as a lysosomal storage disease. Lysosomes are the major digestive units in cells. Enzymes within lysosomes break down or “digest” complex molecules such as mixtures of carbohydrates and fats (like glycosphingolipids). When one of these lysosomal enzymes (such as b-hexosaminidase A) is missing or ineffective, glycosphingolipids start to build up in the lysosome. If there is too much accumulation of these materials in the lysosome, the cells in the nervous system degenerate and die, triggering an inflammatory response that amplifies damage in surrounding tissue.
The most common form of Tay-Sachs disease is the Infantile form, which can present around 6 months of age as reduced vision and an exaggerated startle response and eventually progress to a gradual loss of acquired skills and seizures by age 2 and leads early death, usually by the age of 5. There is also a juvenile version of the disease beginning at about the age of 5 years of age and adult forms of Tay-Sachs disease also known as late-onset Tay Sachs disease (LOTS) beginning in the late teens and beyond. All three forms of Tay-Sachs disease are inherited in an autosomal recessive manner and the age of onset is a function of the amount, if any, of residual enzyme activity.
Infantile Tay-Sachs Disease
The infantile form of Tay-Sachs disease is characterized by complete or almost complete lack of b-hexosaminidase A enzyme activity. The disorder often progresses rapidly, resulting in significant cognitive and physical deterioration.
Infants may appear completely unaffected at birth. Initial symptoms, which usually develop between 3 and 6 months, can include mild muscle weakness, twitching or jerking of muscles (myoclonic jerks) and an exaggerated startle response, such as when there is a sudden or unexpected noise. The startle response may be partly due to an increased sensitivity to sound (acoustic hypersensitivity).
Between six and 10 months, affected infants may fail to gain new motor skills. They may no longer make eye contact and there may be unusual eye movements. They may be listless and irritable. As affected infants age, they may experience slow growth, progressive muscle weakness and diminished muscle tone (hypotonia). Affected infants may also exhibit gradual loss of vision, involuntary muscle spasms (myoclonus), slow, stiff movements (spasticity) and the loss of previously acquired skills (i.e., psychomotor regression) such as crawling or sitting up.
A characteristic symptom of infantile Tay-Sachs disease is the development of a macular “cherry red” spot. This ophthalmological finding emerges from abnormal accumulation of pale undigested material in the macula which contrasts to the thin foveal transparent background exposing the normal rich choroidal vasculature. This characteristic finding occurs is approximately 90% of individuals with infantile Tay-Sachs disease.
As affected infants age, more serious complications may develop including seizures, difficulty swallowing, loss of vision, paralysis and progressive hearing loss. Additional cognitive deficits may include confusion, disorientation and/or deterioration of intellectual abilities. Eventually, infants may become unresponsive to their environment and surroundings. By three to five years of age, life-threatening complications begin to occur such as aspiration pneumonia leading to respiratory failure.
Juvenile (Subacute) Tay-Sachs Disease
The onset of juvenile Tay-Sachs disease can be anywhere between 2 and 10 years of age. One of the first signs is often clumsiness and incoordination. This occurs because affected children have issues controlling their body’s movements (ataxia). Children tend to experience a progressive loss of speech, life skills and intellectual abilities. Affected individuals may or may not develop a cherry-red spot in the eyes. Degeneration of the optic nerves (optic atrophy) may occur. Some children may have retinitis pigmentosa, a progressive loss and degeneration of the cells in the retina where shapes and colors are first encoded. Affected children become less responsive to their environment and surroundings. Life-threatening complications usually occur around 15 years of age.
Late-Onset Tay-Sachs Disease
The presentation and symptoms associated with late-onset Tay-Sachs disease vary greatly. Onset of the disease may vary from the late teens to any time in adulthood. This variability may occur even within affected members of the same family. For example, in each family one person may have symptoms in their 20s, while another reaches into their 60s or 70s with relatively milder minor symptoms. The disorder progresses much slower than the infantile or juvenile forms of the disease.
Initial symptoms associated with late-onset Tay-Sachs disease may include progressive muscle weakness and wasting (neurogenic atrophy), incoordination and clumsiness from cerebellar dysfunction (ataxia) or acute psychiatric presentation. As affected individuals age, muscle twitching (fasciculation), cramps, weakness and muscle wasting progresses affecting preferentially the quadriceps and hip flexor muscles, and later, the triceps muscles. Patients need to lock their knees in hyperextension to be able to stand and support their weight. Failure to do so results in falls and eventually leads to the need for a device to assist with walking. Patients may have tremors and progressive slurred speech (dysarthria). Difficulties swallowing (dysphagia) may emerge late in the disease. Some affected individuals experience acute psychiatric manifestations (mania, acute depression or psychosis) that may require emergency psychiatric care. Over time, cognitive difficulties including executive dysfunction and some memory difficulties might emerge.
Tay-Sachs disease is caused by a change (variant) in the hexosaminidase subunit alpha (HEXA) gene. Genes provide instructions for the basic structure of proteins, all of which play a critical role in many functions and structure of the body. When a variant occurs in a gene, the protein product may be faulty, inefficient or absent. Depending upon the functions of the protein, this can affect many organ systems of the body, including the brain.
The HEXA gene encodes the structure of the HEXA protein which is a subunit of the enzyme hexosaminidase A. More than 80 different variants of the HEXA gene have been identified in individuals with the disease. Inheriting two variant copies of the HEXA gene (homozygotes) causes deficiency of the hexosaminidase A enzyme, which is necessary to breakdown GM2-ganglioside (a glycosphingolipids) within cells of the body. Failure to break down GM2-ganglioside results in its abnormal accumulation in brain and nerve cells eventually resulting in the progressive deterioration of the central nervous system.
In infantile Tay-Sachs disease, there is a complete lack of hexosaminidase A. In juvenile and late-onset Tay-Sachs disease, there is minimal but still some residual hexosaminidase A enzyme activity which explains why these disorders might be less severe and progress at a slower pace than infantile Tay-Sachs disease.
Tay-Sachs disease is inherited as an autosomal recessive disease. Recessive genetic disorders occur when an individual inherits a disease-causing gene variant from each parent. If an individual receives one normal gene and one disease-causing gene variant, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the gene variant and have an affected child is 25% with each pregnancy. The risk of having a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
Tay-Sachs disease affects males and females in equal numbers. Tay-Sachs disease used to be considered a prototypical disease of Jewish people of Ashkenazi descent. Community counselling and carrier screening efforts in these communities have succeeded in reducing disease prevalence to figures roughly equivalent to those of non-Jewish populations. Increased prevalence of Tay Sachs disease is also reported in other ethnic groups including those living in the Cajun community of Louisiana and southeastern Quebec. In the general population, the carrier rate for Tay-Sachs disease is approximately 1 in 250-300 people.
The diagnosis of Tay-Sachs disease may be confirmed by a thorough clinical evaluation and specialized tests such as blood tests that measure the enzyme activity levels of hexosaminidase A. Molecular genetic testing for variants in the HEXA gene can confirm a diagnosis of Tay-Sachs disease. With the advent of more widely available gene panels and exome and whole genome sequencing, more patients are initially diagnosed by molecular testing followed by enzymatic confirmation.
Prenatal diagnosis is possible through molecular genetic testing of tissue samples obtained through chorionic villus sampling (CVS) or amniocentesis if the disease-causing variants in the HEXA gene are known in the family. During amniocentesis, a sample of fluid that surrounds the developing fetus is removed, while CVS involves the removal of tissue samples from a portion of the placenta.
Carrier testing for Tay-Sachs disease can be accomplished from a blood sample and determines whether an individual carries one disease-causing variant of the HEXA gene. Relatives of individuals with Tay-Sachs disease can be tested to determine whether they are carriers. Couples who are planning to have a child and have any Jewish ancestry (not just Ashkenazi) are encouraged to have carrier screening before pregnancy.
For couples that find that they are carriers, there are several options available for starting a family. These options include assisted reproductive technologies (ART) such as in vitro fertilization (IVF) and adoption. Couples are encouraged to consult with a genetic counselor to discuss these options.
Genetic counseling is available to assist families in testing decisions, interpreting test results and understanding health insurance coverage. Families are strongly encouraged to seek out a genetic counselor, particularly before carrier screening, to be most informed when making genetics healthcare decisions. For assistance finding a genetic counselor, visit: https://findageneticcounselor.nsgc.org/?reload=timezone
Treatment
There is no approved treatment for Tay-Sachs disease. Treatment is directed toward individual symptom management. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, neurologists, speech pathologists, specialists who assess and treat hearing problems (audiologists), eye specialists and other health care professionals may need to collaborate to develop a plan for an affected child’s treatment.
Because of the potential for feeding difficulties, infants should be monitored for nutritional status and proper hydration. Nutritional support and supplementation may be necessary. Occasionally, the insertion of a feeding tube may be required to help prevent food, liquid or other foreign material from accidentally going into the lungs (aspiration).
Anticonvulsants may be used to treat seizures in some people with Tay-Sachs disease but may not be effective in all people. The types and frequency of seizures can change over time in some individuals which will require a change in medication type or dosage.
Genetic counseling is recommended for affected individuals and their families. Psychosocial support is recommended for the entire family.
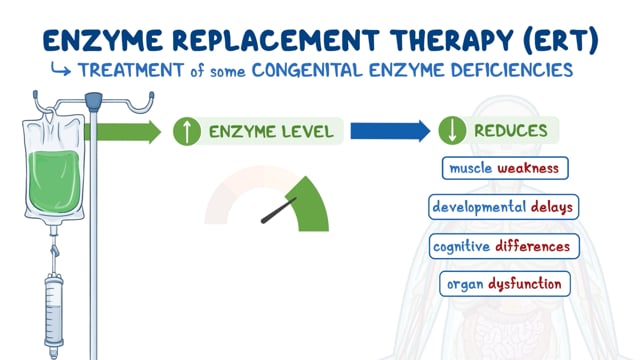
Enzyme replacement therapy (ERT) has been considered and developed for lysosomal storage disorders. In Tay-Sachs disease, enzyme replacement therapy involves replacing a missing or deficient hexosaminidate A. Strategies to deliver the replacement enzyme (a large molecule) to cross the blood-brain barrier are being investigated.
Chaperone therapy is also being studied for the therapy of Tay-Sachs disease. This type of therapy involves very small molecules called chaperones that attach to newly synthetized hexosaminidase A enzymes. This approach seeks to protect degradation of the abnormal hexosaminidase A inside the cells before it is degraded, thus allowing hexosaminidase A, albeit deficient, to have activity for a longer time-period. A chaperone called pyrimethamine has been studied as a treatment for Tay-Sachs disease. Affected individuals who took the medication showed increased laboratory activity of hexosaminidase A enzyme. However, increased activity did not lead to any noticeable improvement in symptoms and was accompanied by undesirable side effects.
Gene therapy is also being studied as another possible approach to therapy for some lysosomal storage disorders. In gene therapy, the defective gene present in a patient is replaced with a normal gene to enable the production of active enzyme and prevent the development and progression of the disease. Some delivery methods for gene therapy results permanent transfer of a normal copy of the HEXA gene leading to synthesis of the active enzyme at all sites of disease, this form of therapy is theoretically most likely to lead to a “cure.”
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government website.
For information about clinical trials being conducted at the National Institutes of Health (NIH) in Bethesda, MD, contact the NIH Patient Recruitment Office:
Toll-free: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website:
https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, contact: www.centerwatch.com
For more information about clinical trials conducted in Europe, contact: https://www.clinicaltrialsregister.eu/
JOURNAL ARTICLES
Liguori M, Tagarelli G, Romeo N, Bagala A, Spadafora P. Identification of a patient affected by “juvenile chronic” Tay Sachs disease in South Italy. Neurol Sci. 2016;37:1883-1885. https://www.ncbi.nlm.nih.gov/pubmed/27351546
Steiner KM, Brenck J, Goericke S, Timmann D. Cerebellar atrophy and muscle weakness: late-onset Tay-Sachs disease outside Jewish populations. BMJ Case Rep. 2016;2016. https://www.ncbi.nlm.nih.gov/pubmed/27033294
Osher E, Fattal-Valevski A, Sagie L, et al. Effect of cyclic, low dose pyrimethamine treatment in patients with late onset Tay Sachs: an open lab el, extended pilot study. Orphanet J Rare Dis. 2015;10:45. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4404274/
Hall P, Minnich S, Teigen C, Raymond K. Diagnosing lysosomal storage disorders: the GM2 gangliosidoses. Curr Protoc Hum Genet. 2014;83:17.161-8. https://www.ncbi.nlm.nih.gov/pubmed/25271840
Smith NJ, Winstone AM, Stellitano L, Cox TM, Verity CM. GM2 gangliosidosis in a UK study of children with progressive neurodegeneration: 73 cases reviewed. Dev Med Child Neurol. 2012;54:176-182. https://www.ncbi.nlm.nih.gov/pubmed/22115551
Osher E, Fattal-Valevski A, Sagie L, et al. Pyrimethamine increases Beta-hexosaminidase A activity in patients with late onset Tay Sachs. Mol Genet Metab. 2011;102:356-363. https://www.ncbi.nlm.nih.gov/pubmed/21185210
Bley AE, Giannikopoulos OA, Hayden D, et al. Natural history of infantile G(M2) gangliosidosis. Pediatrics. 2011;128:e1233-1241. https://www.ncbi.nlm.nih.gov/pubmed/22025593
Clarke JT, Mahuran DJ, Sathe S, et al. An open-label phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants). Mol Genet Metab. 2011;102:6-12. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3019177/
Schneider A, Nakagawa S, Keep R, et al. Population-based Tay-Sachs screening among Ashkenazi Jewish young adults in the 21st century: hexosaminidase A enzyme assay is essential for accurate testing. Am J Med Genet A. 2009;149A:2444-2447. https://www.ncbi.nlm.nih.gov/pubmed/19876898
Shapiro BE, Pastores GM, Gianutsos J, Luzy C, Kolodny Eh. Miglustat in late-onset Tay-Sachs disease: a 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genet Med. 2009;11:425-533. https://www.ncbi.nlm.nih.gov/pubmed/19346952
Shapiro BE, Logigian EL, Kolodny EH, Pastores GM. Late-onset Tay-Sachs disease: the spectrum of peripheral neuropathy in 30 affected patients. Muscle Nerve. 2008;38:1012-1015. https://www.ncbi.nlm.nih.gov/pubmed/18642377
Maegawa GH, Stockley T, Tropak M, et al. The natural history of juvenile or subacute GM2 gangliosidosis: 21 new cases and literature review of 134 previously reported. Pediatrics. 2006;118:el550-562. https://www.ncbi.nlm.nih.gov/pubmed/17015493
Bach G, Tomczak J, Risch N, Ekstein J. Tay-Sachs screening in the Jewish Ashkenazi population: DNA testing is the preferred procedure. Am J Med Genet. 2001;99:70-75. https://www.ncbi.nlm.nih.gov/pubmed/11170098
Hill LW, Schorr SJ. Prenatal screening for Tay-Sachs disease by Louisiana obstetricians: a survey study. South Med J. 2001;94:910-12. https://www.ncbi.nlm.nih.gov/pubmed/11592753
Guidotti JE, Mignon A, Haase G, et al. Adenoviral gene therapy of the Tay-Sachs disease in hexosaminidase A-deficient knock-out mice. Hum Mol Genet. 1999;8:831-38. https://www.ncbi.nlm.nih.gov/pubmed/10196372
Platt FM, Butters TD. New therapeutic prospects for the glycosphingolipid lysosomal storage diseases. Biochem Pharmacol. 1998;56:421-30. https://www.ncbi.nlm.nih.gov/pubmed/9763217
De Gasperi R, Gama Sosa A, Battistini S, et al. Late-onset GM2 gangliosidosis: Ashkenazi Jewish family with an exon 5 mutation (Tyr– >His) in the Hex A alpha-chain gene. Neurology. 1996;47:547-52. https://www.neurology.org/content/47/2/547.short
Nakai H, Byers MG, Nowak SJ, Shows TB. Assignment of beta-hexosaminidase A alpha-subunit to human chromosomal region 15q23 – q24. Cytogenet Cell Genet. 1991;56:164. https://www.ncbi.nlm.nih.gov/pubmed/1829032
Navon R, Kolodny EH, Mitsumoto H, Thomas GH, Proia RL. Ashkenazi-Jewish and non-Jewish adult GM2 gangliosidosis patients share a common genetic defect. Am J Med Genet. 1990;46:817-21. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1683663/
Navon R, Proia RL. The mutations in Ashkenazi Jews with adult GM2 gangliosidosis, the adult form of Tay-Sachs disease. Science. 1989;243:1471-74. https://www.ncbi.nlm.nih.gov/pubmed/2522679
INTERNET
Assisted Reproductive Technologies. Society for Assisted Reproductive Technology (SART) website. Available at: https://www.sart.org/patients/a-patients-guide-to-assisted-reproductive-technology/general-information/assisted-reproductive-technologies/ Accessed Jan 22, 2025.
Toro C, Shirvan L, Tifft C. HEXA Disorders. 1999 Mar 11 [Updated 2020 Oct 1]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1218/ Accessed Jan 22, 2025.
Tay-Sachs Disease. MedlinePlus. Last updated September 30, 2021. Available at: https://ghr.nlm.nih.gov/condition/tay-sachs-disease Accessed Jan 22, 2025.
Tay-Sachs Disease. Online Mendelian Inheritance in Man, OMIM (TM). John Hopkins University, Baltimore, MD. MIM Number:272800; Updated: 06/24/2022. Available at: https://omim.org/entry/272800 Accessed Jan 22, 2025.

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportOnline Mendelian Inheritance In Man (OMIM) has a summary of published research about this condition and includes references from the medical literature. The summary contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. OMIM is authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine.
View reportGeneReviews has an article on this condition covering diagnosis, management, and inheritance. Each article is written by one or more experts on the specific disease and is reviewed by other specialists. The article contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. The GeneReviews database is managed by the University of Washington.
View reportMedlinePlus has information about this condition that may include a description, frequency, causes, inheritance, and links to more information. The information is written for the public, including patients, caregivers and families. MedlinePlus is a service of the National Library of Medicine (NLM), which is part of the National Institutes of Health (NIH).
View reportPlease complete this form to access the requested resource.

