

Last updated:
1/17/2025
Years published: 1984, 1985, 1986, 1987, 1988, 1989, 1990, 1991, 1992, 1994, 1995, 1996. 1997, 1998, 1999, 2000, 2002, 2007, 2009, 2011, 2014, 2017, 2025
NORD gratefully acknowledges Alaina Thorne, Bridget Murphy and Professor Barbara Calhoun, University of Notre Dame and Patrick A. Flume, MD, Distinguished Professor of Medicine and Pediatrics, The Powers-Huggins Chair for Cystic Fibrosis, Medical University of South Carolina, for assistance in the preparation of this report.
Cystic fibrosis (CF) is an autosomal recessive genetic disorder caused by changes (variants) in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that affect the regulation of water and salt flow into and out of cells.1 CF affects the exocrine glands, leading to the production of abnormally thick mucus. This mucus clogs essential organs such as the lungs, pancreas and intestines, causing inflammation, obstruction and frequent infections.2 CF affects multiple organ systems, with common symptoms including the following:
Historically, lung (pulmonary) disease has been the major cause of health problems and death related to CF. Over time, CF causes progressive lung damage, leading to scarring of the airways, loss of lung function and eventual respiratory failure. Research identifying the structure and function of the CFTR gene has helped develop drugs that treat the basic defect, modulating CFTR to increase the quantity of the protein produced by the gene and how well it functions. These drugs work for only specific CFTR gene variants but could improve the health of most people with CF. Nearly 90% of people with CF have a variant that is responsive to these medications, although not all are able to tolerate the treatment.1
A newborn screening program in the U.S. allows for early diagnosis, which has significantly reduced the number of people identified later in life, however, people still can be diagnosed in adulthood.3 The incidence of CF is estimated to be 1/4,000 births in the United States.4
Cystic fibrosis (CF) causes mucus in the body to become thicker and stickier than normal, which can block vital passageways and tubes in the body. These blockages primarily affect the lungs and pancreas, but they can also impact on the intestines, liver, sweat glands, and reproductive organs.5
The symptoms of CF can vary widely between individuals. Some people may only have mild respiratory issues, while others experience severe complications in multiple organs. Not everyone with CF will have all the symptoms discussed below. 2,6
Respiratory problems 7,8,9
Many people with CF experience breathing problems due to mucus blocking their airways. These blockages can lead to more serious lung diseases over time. Possible symptoms and complications include:
Pancreatic problems7
Most people with CF have pancreatic insufficiency, meaning their pancreas cannot produce enough enzymes to digest food or hormones like insulin to regulate blood sugar
Gastrointestinal problems10,11
CF can also affect the digestive system causing:
People with CF may also have a higher risk of:7
Additional symptoms include:12
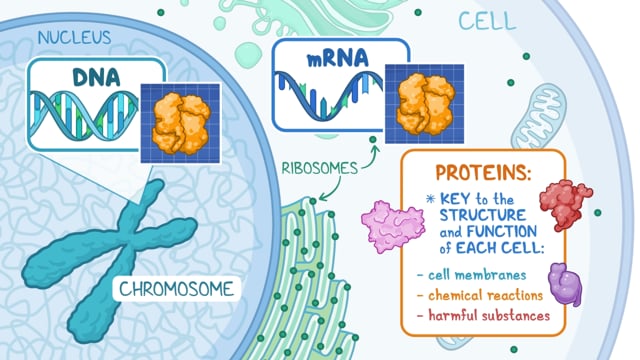
Cystic fibrosis is caused by changes (variants) of the CFTR gene.
The CFTR gene is primarily active (expressed) in cells lining the airways, gastrointestinal tract, pancreas, sweat glands and genitourinary system. The gene controls the production of a protein that regulates the transfer of chloride and sodium across cell membranes. Without chloride, sodium cannot form salt water. In many organs, this leads to abnormally thick secretions (mucus), leading to tissue damage. This thick mucus in the lungs allows bacteria to accumulate, leading to inflammation and pulmonary damage. In the pancreas, thick secretions block the ducts that allow pancreatic enzymes to enter the digestive system, creating pancreatic insufficiency. This blockage of ducts affects the function of many organs (liver disease, infertility/subfertility, bowel obstruction, pansinusitis).7
There are over 700 disease-causing CFTR variants identified resulting in a large range of protein dysfunction and variable signs and symptoms of the disease. The most common variants are classified into 6 groups or classes. Classes I, II, and III result in little to no CFTR protein function and are often associated with more severe disease, whereas classes VI, V, and IV result in some CFTR protein function and milder disease.1
CF is inherited in an autosomal recessive pattern. Recessive genetic disorders occur when an individual inherits a disease-causing gene variant from each parent. If an individual receives one normal gene and one disease-causing gene variant, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the gene variant and have an affected child is 25% with each pregnancy. The risk of having a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.5
CF affects at least 100,000 individuals worldwide, with 40,000 individuals in the US.7 One thousand new cases are diagnosed each year, with males and females affected in equal numbers. The disorder occurs predominantly among Caucasians but is clearly affecting other races. CF occurs in many different ethnicities and should not be limited to Caucasians in consideration. CF occurs in approximately one of every 3,200 live Caucasian births compared to one in 3,900 live births of all Americans. Although incidences of CF are increased in several populations (ex: Amish, Ashkenazi Jewish, etc.), CF occurs in individuals of all ethnicities. CF affects individuals of all ages; with improved survival associated with treatment now more than 50% of people with CF in the US are adults (i.e. older than age 18 years). In many cases, CF is apparent soon after birth, and most affected individuals are diagnosed by age 3. Most people with CF are now identified at birth through newborn screening, but there are still adults being diagnosed.6
A diagnosis of cystic fibrosis may be suspected based on newborn screening, identification of characteristic symptoms (e.g., pulmonary disease, pancreatic insufficiency) or a positive family history. 3,11 All 50 states have newborn screening for CF. In most states, immunoreactive trypsinogen (IRT) assays are performed on dried blood spots from newborns. The IRT assays measure the amount of trypsinogen in a person’s blood. IRT assays are used as a screening test for cystic fibrosis (CF) in newborns and to diagnose pancreatic problems in children and adults. Trypsinogen is synthesized in the pancreas and high IRT levels are typically seen in CF. People who have abnormal IRT results must be followed up with sweat testing and/or molecular genetic (DNA-based) testing.16,3
Sweat testing is a painless and simple procedure that measures the amount of salt in the sweat.3 Elevated chloride level results (≥60 mEq/L) establish a diagnosis of CF. Intermediate chloride level results (30-59 mEq/L) require nasal potential difference (NPD) testing1. The NPD test measures how well the cells in the nasal passage are moving ions (charged particles), which can be used to diagnose CF as people with CF often have an abnormal test due to a faulty protein that regulates ion movement.
After diagnosis, the parents of the affected person should receive molecular genetic testing to confirm that both parents are carriers of a CFTR variant.
Cystic fibrosis (CF) is a complex condition that requires individualized and tailored treatments to address its impact on different parts of the body. While there is no cure, the treatment focuses on reducing thick mucus in the lungs, preventing infections, managing digestive issues and supporting overall health.5 Treatment may include:
In severe cases, lung transplantation may be an option for people with advanced-stage lung disease. This option requires careful consideration of risks and benefits and long-term commitment to anti-rejection medications. 3, 20
Because CF affects many systems, patients are cared for by a team of specialists, including pulmonologists, gastroenterologists, nutritionists, physical therapists and genetic counselors as well as mental health and fertility specialists, as needed. 21 Working with a CF care team can help affected people manage their symptoms, prevent complications and have healthier lives.
Patients and families should consult genetic counselors to help them understand CF inheritance and assess risks for future children.6
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. Government funding, and some supported by private industry, are posted on this government website.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Tollfree: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website:
https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, contact:
www.centerwatch.com
For information about clinical trials conducted in Europe, contact:
https://www.clinicaltrialsregister.eu/

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportOnline Mendelian Inheritance In Man (OMIM) has a summary of published research about this condition and includes references from the medical literature. The summary contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. OMIM is authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine.
View reportGeneReviews has an article on this condition covering diagnosis, management, and inheritance. Each article is written by one or more experts on the specific disease and is reviewed by other specialists. The article contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. The GeneReviews database is managed by the University of Washington.
View reportMedlinePlus has information about this condition that may include a description, frequency, causes, inheritance, and links to more information. The information is written for the public, including patients, caregivers and families. MedlinePlus is a service of the National Library of Medicine (NLM), which is part of the National Institutes of Health (NIH).
View reportPlease complete this form to access the requested resource.

