

Last updated:
December 05, 2017
Years published: 1986, 1987, 1988, 1990, 1994, 1995, 2003, 2017
NORD gratefully acknowledges Shunji Tomatsu, MD, PhD, Director, Skeletal Dysplasia Lab, Departments of Biomedical Research and Orthopedics, Nemours/Alfred I. duPont Hospital for Children; Professor, Department of Pediatrics, Thomas Jefferson University, for assistance in the preparation of this report.
Summary
Mucopolysaccharidosis type VII (MPS VII) is a rare, progressive inborn error of metabolism that affects many parts of the body. The clinical features of MPS VII vary from patient to patient, but all have short stature due to growth retardation and skeletal abnormalities, changes in bones visible on X-rays (dysostosis multiplex), and some degree of intellectual disability. Survival into adulthood is common for patients with milder cases and osteoarthritis is a common complication. MPS VII is inherited as an autosomal recessive genetic condition.
Introduction
MPS VII is in a group of hereditary metabolic diseases known as the mucopolysaccharidoses which in turn, are part of a group known as lysosomal storage disorders. Lysosomes function as the primary digestive units within cells. Enzymes within lysosomes break down or digest particular nutrients, such as certain carbohydrates and fats. In individuals with MPS disorders, including MPS VII, deficiency or improper functioning of lysosomal enzymes leads to an abnormal accumulation of certain complex carbohydrates in cells (mucopolysaccharides, also known as glycosaminoglycans) within various tissues, such as the skeleton, joints, brain, spinal cord, heart, spleen, or liver.
The most severe cases of MPS VII are characterized by hydrops fetalis, or when excess fluid accumulates in the body before birth. This can result in a stillborn or death shortly after birth. Neonatal jaundice or the yellowing of the skin may occur.
Children with more mild cases of MPS VII begin to show symptoms in early childhood. Children with MPS VII may have an unusually short trunk and growth disability, resulting in short stature (short trunk dwarfism). The head may be excessively large (macrocephalic) and the neck may be short. A variety of multiple bone deformities (dysostosis multiplex), which are frequently observed in people with mucopolysaccharidoses, are also common in children with MPS VII. These bone deformities may include a prominent breast bone (pectus carinatum), flared ribs, frequent hip dislocations, “frozen” joints (contractures), club foot, and/or an inward curve of the knees and outward bowing of the ankles (genu valgum).Rarely, spinal malformations may be present including mild curvature of the spine from side to side (scoliosis) and/or front to back (kyphosis). Children with this disorder may develop an unusual “coarse” facial appearance. Between the ages of 7 months and 8 years, cloudiness (opacity) may occur in the corneas of the eyes.
Affected individuals may have developmental delays in language and speech and progressive intellectual disability, although intelligence is normal in some people with this condition.
Other symptoms of MPS VII may include a swollen abdomen due to abnormal enlargement of the liver and/or spleen (hepatosplenomegaly) and protrusion of the intestines through an abnormal opening in the muscular wall of the abdomen (inguinal hernia). In some children, the intestines may also protrude through the abdominal wall in the area of the navel (umbilical hernia). Some affected children may experience profound hearing loss, recurrent upper respiratory and middle ear infections, thickening of the soft tissues of the throat and/or vocal cords, an abnormally enlarged tongue (macroglossia), and/or heart problems (i.e., heart murmur or aortic regurgitation). Excessive hairiness (hirsutism) may be present.
Children may develop carpal tunnel syndrome characterized by numbness, tingling and weakness in the hands and fingers.
Survival to age 19-20 years has been reported in mild cases. Life expectancy is reduced as a result of frequent upper respiratory tract infections, neurodegenerative complications and abnormalities of the gastrointestinal tract.
A mild form of MPS VII, beginning during the 2nd decade of life, has been described in the medical literature. In these patients, the symptoms of the disorder appear to be less severe than classical MPS VII and may include minor bony changes and very mild facial coarseness. Growth rate and mental abilities are usually normal. Abnormal enlargement of the liver and spleen has not been noticed in this form of MPS VII.
MPS VII is caused by changes (mutations) in the GUSB gene that lead to deficiency of the beta-glucuronidase enzyme. A variety of different mutations in this gene may account for the wide range of symptoms and physical findings as well as the variability in the age of onset.
The beta-glucuronidase enzyme is involved in the breakdown of large sugar molecules called glycosaminoglycans or GAGs. The shortage of beta-glucuronidase leads to a buildup of GAGs in the cells, specifically in the lysosomes. Lysosomes digest and recycle molecules; this disorder causes a lysosomal storage disorder. The accumulation of GAGs increases the size of the lysosomes causing the tissues and organs to become enlarged. GAGs are believed to interpret the function of proteins within the lysosomes and disrupt the normal function of cells.
MPS VII is inherited as an autosomal recessive genetic condition. Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
MPS VII is extremely rare, affecting only about one in 250,000 births. Fewer than 100 cases have been reported in the United States. Males and females are affected in equal numbers.
Urinary levels of the mucopolysaccharides (dermatan sulfate, heparan sulfate, and chondroitin sulfate) are increased in affected individuals. The diagnosis of MPS VII may be confirmed by a thorough clinical evaluation that includes a detailed patient history and specialized tests that measure the level of beta-glucuronidase activity in blood or skin cells. Molecular genetic testing for mutations in the GUSB gene is available to confirm the diagnosis. Prenatal diagnosis is possible through amniocentesis or chorionic villus sampling to measure beta-glucuronidase activity or molecular genetic testing for GUSB gene mutations.
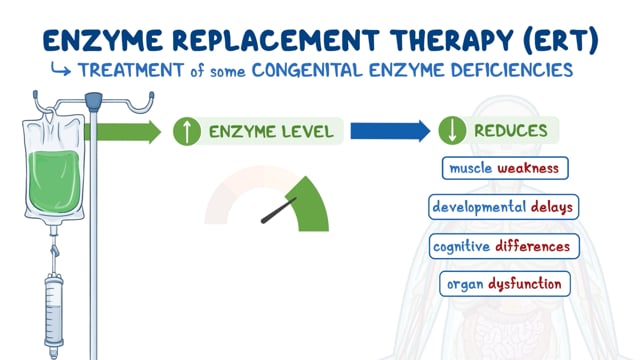
Treatment
In 2017, Mepsevii (vestronidase alfa-jvbk), an enzyme replacement therapy, was approved to treat pediatric and adult patients with MPS VII. Mepsevii is manufactured by Ultragenyx Pharmaceutical, Inc.
Other treatment of MPS VII is symptomatic and supportive. Bone deformities and hernias may require surgical correction. Ocular and cardiovascular abnormalities may also be treated surgically. Patients with MSP disorders may be sensitive to anesthesia because of malformations in the airway or cervical spine; therefore, precautions should be taken prior to surgery. Genetic counseling is recommended for people with MPS VII and their families.
Hematopoietic Stem Cell Therapy (HSCT)
A Japanese patient underwent successful HSCT at 12 years of age and after 2 years the levels of GUS were stable, with improvement of motor functions (walking, riding, and taking bath alone), recurrent infections and snoring; neurological damage has been kept stable (Yamada et al., 1998). A Mexican female patient with MPS VII with genotype p.P408S/p.P415L underwent HSCT at 11 years of age and was reported to be doing well 2 years after the transplantation (Islam et al., 1996). There were more than 7 patients that underwent HSCT (Islam et al., 1996; Yamada et al., 1998; Montaño et al., 2016). The limited results suggest that HSCT can slow or even prevent further neurological complications, but has little to no effect on the skeletal disease unless it is performed in an early stage.
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov . All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Toll free: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website:
https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, contact:
www.centerwatch.com
For information about clinical trials conducted in Europe, contact:
https://www.clinicaltrialsregister.eu/
TEXTBOOKS
Clarke JTR. MPS-VII. In: The NORD Guide to Rare Disorders, Philadelphia: Lippincott, Williams and Wilkins, 2003:481.
Beighton P, McKusick VA. Heritable Disorders of Connective Tissue, 5th ed. St. Louis: CV Mosby, 1993.
JOURNAL ARTICLES
Montaño, AM, Lock-Hock, N, Steiner, RD, et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII). J. Med. Genet. 2016;53, 403-418.
Fox, JE, Volpe, L, Bullaro, J, Kakkis, ED, Sly, WS First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol. Genet. Metab. 2015;114, 203-8.
Yamada, Y, Kato, K, Sukegawa, K, Tomatsu, S, Fukuda, S, Emura, S, Kojima, S, Matsuyama, T, Sly, WS, Kondo, N, Orii, T. Treatment of MPS VII (Sly disease) by allogenic BMT in a female with homozygous A619V mutation. Bone Marrow Transplant. 1998;21:629-634
Islam, MR, Vervoort, R, Lissens, W, Hoo, JJ, Valentino, LA, Sly, WS. Beta-Glucuronidase P408S, P415L mutations: evidence that both mutations combine to produce an MPS VII allele in certain Mexican patients. Hum. Genet. 1996;98, 281-4.
Kagie MJ, et al. Beta-glucuronidase deficiency as a cause of fetal hydrops. Am J Med Genet.1992;42(5):693-5.
De Kramer RD, et al. Mucopolysaccharidosis type VII (beta-glucuronidase deficiency): A chronic variant with an oligosymptomatic severe skeletal dysplasia. Am J Med Genet. 1992;44(2):145-52.
Peterson L, et al. Mucopolysaccharidosis type VII. A morphoplogic, cytochemical, and ultrastructural study of the blood and bone marrow. Am J Clin Pathol. 1992;78(4):544-48.
Kjellen L, Lindahl V. The proteoglycans structures and functions. Ann Rev Biochem. 1991:60:443-475.
Tomatsu S, et al. Mucopolysaccharidosis type VII: Characterization of mutations and molecular heterogeneity. Am J Hum Genet. 1991;48(1):89-96.
INTERNET
McKusick VA, ed. Online Mendelian Inheritance in Man (OMIM). Baltimore, MD: The Johns Hopkins University; Entry No. 253220; Last Update: 02/12/2014. https://omim.org/entry/253220 Accessed January 17, 2017.
Banikazemi, Maryam. Genetics of Mucopolysaccharidosis Type VII. Medscape. Last Update November 10, 2015. https://emedicine.medscape.com/article/944298-overview#a4 Accessed January 17, 2017.
Genetics Home Reference. Mucopolysaccharidosis type VII. August, 2010. https://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-vii Accessed January 17, 2017.
National MPS Society. A Guide to Understanding MPS VII. 2008. https://www.mpssociety.org/wp-content/uploads/2011/07/MPS_VII_2008.pdf Accessed January 17, 2017.
Froissart Roseline, Maire Irene. Mucopolysaccharidosis type VII. Orphanet. Last Update February 2007. https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=40&Disease_Disease_Search_diseaseGroup=Mucopolysaccharidosis-Type-VII&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Mucopolysaccharidosis-type-7&title=Mucopolysaccharidosis-type-7&search=Disease_Search_Simple Accessed January 17, 2017.

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportOnline Mendelian Inheritance In Man (OMIM) has a summary of published research about this condition and includes references from the medical literature. The summary contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. OMIM is authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine.
View reportGeneReviews has an article on this condition covering diagnosis, management, and inheritance. Each article is written by one or more experts on the specific disease and is reviewed by other specialists. The article contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. The GeneReviews database is managed by the University of Washington.
View reportMedlinePlus has information about this condition that may include a description, frequency, causes, inheritance, and links to more information. The information is written for the public, including patients, caregivers and families. MedlinePlus is a service of the National Library of Medicine (NLM), which is part of the National Institutes of Health (NIH).
View reportPlease complete this form to access the requested resource.

