

Last updated:
April 22, 2019
Years published: 1986, 1987, 1988, 1990, 1991, 1995, 1997, 1998, 1999, 2000, 2001, 2002, 2003, 2004, 2007, 2019
NORD gratefully acknowledges Lorne A. Clarke MD, CM, FRCPC, FCCMG, Professor Medical Genetics, University of British Columbia, Clinical and Biochemical Geneticist, Provincial Medical Genetics Program, for assistance in the preparation of this report.
Summary
Mucopolysaccharidosis type I (MPS I) is a rare genetic disorder that affects many parts of the body (multisystem). Children with MPS I are described as having either a severe or attenuated (meaning reduced) form of the disorder based on age of onset, severity of symptoms, rate of disease progression and whether there is early and direct involvement of the brain. The determination of whether an individual has severe or attenuated MPS I is critical as different treatment options are available. Individuals with severe MPS I usually have symptoms apparent by 6 months of age whereas individuals with attenuated MPS I may not become apparent until after the age of 3 years with many attenuated MPS I individuals not diagnosed until late childhood or even the teenage years. Individuals with severe MPS I have onset of symptoms in early infancy with evidence of early progressive intellectual decline and when untreated, die within the first decade. In contrast, individuals with attenuated MPS I have later onset of disease symptoms, more slowly progressive disease with sparing of intelligence and can have a near normal life expectancy. MPS I is caused by variations (AKA mutations or pathogenic sequence variants) in the IDUA gene and is inherited in an autosomal recessive pattern. Therefore, both parents of every affected MPS I individual are carriers of MPS I. Being a carrier for MPS I does not lead to symptoms.
Introduction
MPS I is member of a group of hereditary metabolic diseases known as the mucopolysaccharidoses which, in turn, are part of a larger group of diseases known as lysosomal storage disorders (LSDs). Lysosomes function as the primary digestive and recycling units within cells. Enzymes within lysosomes break down or digest particular cellular components, such carbohydrates, proteins and fats to their basic units which can then be recycled. Healthy cells and organs are constantly breaking down, recycling and building new cellular components. In individuals with MPS disorders, including MPS I, deficiency or improper functioning of lysosomal enzymes leads to an abnormal accumulation of a particular complex carbohydrate known as glycosaminoglycans. Glycosaminoglycans were once known as mucopolysaccharides and are how these disorders got their names. When cells cannot breakdown these glycosaminoglycans they then accumulate within various tissues, such as the bones, joints, brain, spinal cord, heart, spleen, or liver and lead to the symptoms that MPS I individuals have.
MPS I is best thought of as a spectrum of disease that ranges from severe forms (Hurler syndrome) that are present very early in life to less severe forms that may not become apparent until much later in childhood. Individuals with MPS I were previously classified as having either a severe, mild, or intermediate form of the disorder. The severe form was known as Hurler syndrome, the mild form was known as Scheie syndrome, and the intermediate form was known as Hurler-Scheie syndrome. Although the term Hurler syndrome is still used, the term attenuated MPS I is now used in place of Hurler-Scheie and Scheie.
The specific signs and symptoms seen in MPS I are highly variable and depends on numerous factors including what form of MPS I a child has, when treatment was first started, and how an individual child responds to various treatment options. It is important to note that MPS I is a progressive disorder and therefore the symptoms that a child has are also related to the age of the child at the time of diagnosis. The age of onset, disease severity, degree of intellectual disability, and rate of progression vary significantly among affected individuals.
Severe Mucopolysaccharidosis Type I
Common signs and symptoms of the severe form of mucopolysaccharidosis type I are delays in the developmental of motor skills and intelligence, and skeletal deformities. Symptoms may not be present at birth and may not become apparent until several months to 1 year of age.
Sometimes, affected infants have an inguinal or umbilical hernia. Inguinal hernia is when some abdominal tissue or part of the small intestine pushes through a bulge or tear in the abdominal muscles near the groin. An umbilical hernia is when some abdominal tissue or part of the small intestine pushes through a bulge or tear in the bellybutton. Affected infants often experience repeated upper respiratory tract infections in the first year of life. Because both hernias and ear infections occur commonly in children, these findings alone do not often lead to diagnosis.
Within the first year to 18 months of age, affected children usually
The signs listed above represent the early signs of severe disease but because this disorder is progressive other potential symptoms occur as the disease progresses and children age and include:
Attenuated Mucopolysaccharidosis Type I
Children described as having the attenuated form of MPS I experience similar signs and symptoms seen in severely affected patients but tend to show slower disease progression and later age of onset of symptoms. A key difference between severe and attenuated patients is that attenuated patients do not show early developmental delay and do not experience progressive decline in mental capabilities. As a group, attenuated patients show significant variability in the symptoms they have and the rate of progression. Some children are mildly affected and have a near normal lifespan, while others develop symptoms during childhood, usually around 6 or 7 years old and can develop life-threatening complications by their teen-aged years or during their 20s. These symptoms of the attenuated form are as described as above for the severe form. The symptoms can be as significant as the severe form, or can be milder. Individuals with attenuated MPS I can have their quality of life significantly affected.
Intelligence is usually not affected in children with attenuated MPS I, but some children and young adults may experience learning disabilities. Affected children will have varying degrees of growth deficiency. They can develop an enlarged liver (hepatomegaly), clouding of the cornea, and heart valve abnormalities during their teen-aged years. Corneal clouding can lead to significant problems with vision. Other eye abnormalities including glaucoma, optic atrophy, and degeneration of the retina can also occur. Progressive heart valve problems can begin as early 10 or 11 years of age. Coronary heart disease can also occur.
Dysostosis multiplex, skeletal malformations, carpal tunnel syndrome, and progressive joint disease can also occur in the attenuated forms. These signs and symptoms are the same as described above for the severe form. Affected individuals may also have a high arch to the feet (pes cavus) and knees that are angled in so that they rub together when the legs are straight (knock knees or genu valgum). Some children may walk on the balls of their feet so that the heels do not touch the ground (toe-walking).
Moderate to severe hearing loss can also occur in the attenuated forms. Some children may develop sleep apnea. Hernias can also be present. Progressive compression of the spinal cord can also be possible and can cause problems including exercise intolerance and reduced activity.
Mucopolysaccharidosis type I is caused by a variation (mutation, pathogenic sequence variant) in the alpha-L-iduronidase (IDUA) gene. Genes provide instructions for creating proteins that play a critical role in many functions of the body. When a mutation of a gene occurs, the protein product may be faulty, inefficient, absent, or overproduced. Depending upon the functions of the particular protein, this can affect many organ systems of the body, including the brain.
The IDUA gene regulates the production of the alpha-L-iduronidase enzyme. This enzyme is needed to break down complex carbohydrates produced in the body called glycosaminoglycans (which used to be called mucopolysaccharides). When the IDUA gene is altered, there are deficient levels of functional alpha-L-iduronidase enzyme. Without proper levels of this enzyme, these glycosaminoglycans, especially dermatan sulfate and heparan sulfate, build up in the lysosomes of all cells. This abnormal accumulation interferes with the proper functions and health of the cells and, ultimately, this leads to progressive damage of the tissues and results in symptoms. In severe MPS I there is a complete absence of the alpha-L-iduronidase enzyme, while with the attenuated forms there is likely a very, very, small amount of enzyme made in the cells.
Genetic diseases are determined by the combination of genes that we receive from our father and mother. We all have 2 copies of the IDUA gene; one we have inherited from our father and one we have inherited from our mother. Disorders inherited in a recessive manner occur when an individual has 2 copies of a gene and both copies have errors in them. This is the case for all MPS I individuals; they have 2 copies of the IDUA gene that are altered; they have received one from each of their parents. The parents are therefore carriers of MPS I. Carriers have one normal copy and one altered copy of the IDUA gene. Carriers are healthy and being a carrier for MPS I will not lead to symptoms. The risk for two carrier parents to both pass the altered IDUA gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
Mucopolysaccharidosis type I affects males and females in equal numbers, with an incidence of about 1 in 100,000 live births for the severe type, and an incidence of about 1in 500,000 live births for the attenuated type. Incidence is the number of people who develop a disorder over a given period of time (e.g. one year). The incidence for MPS disorders collectively is about 1 in 25,000. However, rare disorders, especially milder forms of MPS, often go misdiagnosed or undiagnosed, making it difficult to determine the true frequency in the general population.
A diagnosis of mucopolysaccharidosis type I is based upon identification of characteristic symptoms, a detailed patient and family history, a thorough clinical evaluation and a variety of specialized tests. A diagnosis may be suspected in infants with characteristic early signs. Currently newborn screening for MPS I is being implemented in many areas.
Clinical Testing and Workup
A specialized examination of urine of people with MPS I can reveal elevated levels of glycosaminoglycans (mucopolysaccharides), specifically heparan and dermatan sulfate. This is not diagnostic of MPS I, but is indicative of a mucopolysaccharidosis disorder.
The definitive diagnosis of MPS I requires testing specific cells such as white blood cells (leukocytes) or connective tissue cells (fibroblasts). These tests demonstrate low activity of the alpha-L-iduronidase enzyme.
Molecular genetic testing is often used to confirm a diagnosis. Molecular genetic testing can detect disease-causing alterations (mutations) in the IDUA gene known to cause MPS I, but is available only as a diagnostic service at specialized laboratories.
In the United States, newborn screening for MPS I has recently been approved for inclusion in the Recommended Universal Screening Panel, although each state decides independently when or if, it will be added to its newborn screening panel. One issue with newborn screening for MPS I is that in some cases doctors may not be able to distinguish whether a newborn with MPS I will develop a severe or attenuated form of the disorder. Because the treatments for these forms are different, this can lead to challenges with determining the proper course of treatment. Research is currently underway to look for methods to distinguish infants with severe MPS I from infants with attenuated MPS I.
Treatment
There are three components in the treatment of MPS I:
1) replacing the enzyme that is missing
2) alleviating specific symptoms of disease
3) providing the family with genetic counseling
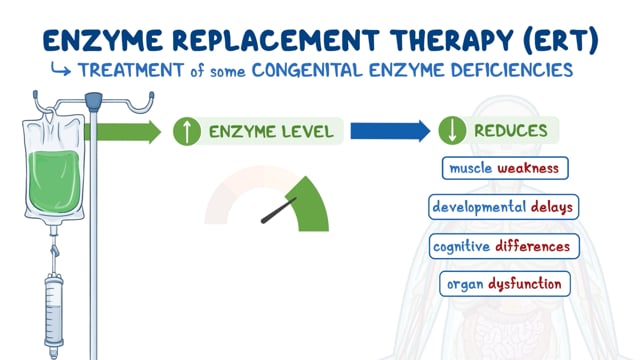
1) Replacing the deficient enzyme: Lysosomal enzymes are unique proteins by virtue of the fact that they can be taken up by cells and used. As such, one potential way to treat MPS I is to give patients the alpha-L-iduronidase enzyme they are missing. This can be done in 2 ways. One way is to infuse them with purified enzyme also known as enzyme replacement therapy (ERT). Enzyme replacement therapy involves replacing the missing enzyme, alpha-L-iduronidase, with a genetically engineered (recombinant) form. This is performed by infusing the enzyme into a patient weekly through an intravenous. In 2003, the U.S. Food and Drug Administration (FDA) approved laronidase (Aldurazyme) for the treatment of most individuals with MPS I. Aldurazyme does not help with symptoms related to damage to the brain and central nervous system because the medication cannot cross the blood-brain barrier, which is the protective network of blood vessels and cells that allow some materials to enter the brain, while keeping other materials out.
Another method to replace the enzyme in patients is to perform hematopoietic stem cell (HSCT) or bone marrow transplantation. Hematopoietic stem cells are specialized cells found in the bone marrow (the soft spongy material found in long bones). These blood stem cells grow and eventually develop into one of the three main types of blood cells– red blood cells, white blood cells or platelets. A transplant is done to replace the bone marrow of an affected individual with marrow from a person who does not have MPS I. The healthy cells produced by the transplant contain sufficient levels of white blood cells that produce alpha-L-iduronidase and these cells then replace in the enzyme deficiency in the tissues. The procedure is expensive and carries the risk of serious complications including death, graft-versus-host disease and other long-term and late effects. HSCT is considered the standard of care for severe MPS I as it is the only treatment currently available that has shown to lead to improved intellectual outcomes in severe MPS I individuals.
Although a HSCT and enzyme replacement therapy can dramatically improve the clinical course they do not cure this disorder and, therefore, many affected individuals still face significant challenges and issues as they age. Generally, the earlier treatment is started the better the outcome and the severity of the disorder and a child’s age often influence the outcome of the procedure. The impact of a HSCT on intellectual disability can vary. Usually, once significant intellectual disability is present, it cannot be reversed, although cognitive decline can be alleviated.
2) Alleviating specific symptoms of disease: An important component of the treatment of mucopolysaccharidosis type I is directed toward the specific symptoms that are apparent in each individual. This requires the coordinated efforts of a team of specialists and include.
Pediatricians
3) Genetic counseling is recommended for affected individuals and their families. Psychosocial support for the entire family is essential as well.
Several of the organizations listed in the Resources section provide support and information on MPS disorders like MPS type I.
There are extensive efforts ongoing to improve the outcomes for patients with MPS I by providing better ways to replace the missing enzyme. These efforts include different forms of enzymes that may be able to access tissues better and gene-based therapies that could replace the missing enzyme. In gene therapy, the defective gene in a patient is either corrected or a normal functioning gene is given to the patient. Therefore, the patient is then able to produce the active enzyme thus preventing the development and progression of the disease. This form of therapy is theoretically most likely to lead to a “cure.” However, at this time, there remain some technical challenges to resolve before gene therapy can be advocated as a viable alternative approach.
Information on current clinical trials is posted on the Internet at https://clinicaltrials.gov/. All studies receiving U.S. Government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Toll-free: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website:
https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, contact:
https://www.centerwatch.com/
For information about clinical trials conducted in Europe, contact:
https://www.clinicaltrialsregister.eu/
JOURNAL ARTICLES
Kuiper GA, Langereis EJ, Breyer S, et al. Treatment of thoracolumbar kyphosis in patients with mucopolysaccharidosis type I: results of an international consensus procedure. Orphanet J Rare Dis. 2019;14:17. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6339313/
Donati MA, Pasquini E, Spada M, Polo G, Burlina A. Newborn screening in mucopolysaccharidoses. Ital J Pediatr. 2018;44(Suppl 2):126. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6238254/
Grosse SD, Lam WKK, Wiggins LD, Kemper AR. Cognitive outcomes and age of detection of severe mucopolysaccharidosis type 1. Genet Med. 2017;19:957-982. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5763496/
Parini R, Deodato F, Di Rocco M, et al. Open issues in mucopolysaccharidosis type I-Hurler. Orphanet J Rare Dis. 2017;12:112. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5472858/
Khan SA, Peracha H, Ballhausen D, et al. Molecular genetics and metabolism. Mol Genet Metab. 2017;121:227-240. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5653283/
Peake RWA, Bodamer OA. Newborn screening for lysosomal storage disorders. J Pediatr Genet. 2017;6:51-60. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5288002/
Kiely BT, Kohler JL, Coletti HY, Poe MD, Escolar ML. Early disease progression of Hurler syndrome. Orphanet J Rare Dis. 2017;12:32. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5307824/
Laraway S, Mercer J, Jameson E, et al. Outcomes of long-term treatment with laronidase in patients with mucopolysaccharidosis type I. J Pediatr. 2016;178:219-226. https://www.sciencedirect.com/science/article/pii/S0022347616307004
Kunin-Batson AS, Shapiro EG, Rudser KD, et al. Long-term cognitive and functional outcomes in children with mucopolysaccharidosis (MPS)-IH (Hurler syndrome) treated with hematopoietic cell transplantation. JIMD Rep. 2016;29:95-102. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5059216/
Khalid O, Vera MU, Gordts PL, et al. Immune-mediated inflammation may contribute to the pathogenesis of cardiovascular disease in mucopolysaccharidosis type I. PLoS One. 2016;11:e0150850. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4795702/
Aldenhoven M, Wynn RF, Orchard PJ, et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125:2164-2172. https://www.bloodjournal.org/content/125/13/2164
Wraith JE, Jones S. Mucopolysaccharidosis type I. Pediatr Endocrinol Rev. 2014;12 Sup 1:102-106. https://www.ncbi.nlm.nih.gov/pubmed/25345091
Wilkinson FL, Holley RJ, Langford-Smith KJ, et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS One. 2012;7:e35787. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3338781/
Braunlin EA, Harmatz PR, Scarpa M, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34:1183-1197. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3228957/
De Ru MH, Boelens JJ, Das AM, et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet J Rare Dis. 2011;6:55. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3170181/
Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology (Oxford). 2011;50 Suppl 5:v19-25. https://academic.oup.com/rheumatology/article/50/suppl_5/v19/1780256
Muenzer J, Wraith JE, Clarke LA, et al. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123:19-29. https://www.ncbi.nlm.nih.gov/pubmed/19117856
INTERNET
Clarke LA. Mucopolysaccharidosis Type I. 2002 Oct 31 [Updated 2016 Feb 11]. In: Pagon RA, Bird TD, Dolan CR, et al., GeneReviews. Internet. Seattle, WA: University of Washington, Seattle; 1993-. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1162/ Accessed February 18, 2019.
Genetic and Rare Diseases Information Center. Mucopolysaccharidosis Type I. December 2012. Available at: https://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-i Accessed February 15, 2019.
Beck M. Mucopolysaccharidosis Type 1. Orphanet Encyclopedia, October 2011. Available at: https://www.orpha.net/consor4.01/www/cgi-bin/OC_Exp.php?lng=EN&Expert=579 Accessed February 15, 2019.

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportPlease complete this form to access the requested resource.

