

Last updated:
September 06, 2019
Years published: 1986, 1987, 1988, 1990, 1999, 2000, 2001, 2002, 2004, 2006, 2007, 2016, 2019
NORD gratefully acknowledges Niraja Suresh, NORD Editorial Intern from the University of Notre Dame, and Maurizio Scarpa, MD, PhD, Regional Coordinating Center for Rare Disease, Udine University Hospital, Udine, Italy, for assistance in the preparation of this report.
Summary
Mucopolysaccharidosis type II (MPS II) is a rare lysosomal inborn error of metabolism that affects every organ of the body. Although the age of onset, disease severity and the rate of progression of the disease vary significantly, initial symptoms and findings associated with MPS II usually become apparent in children from two to four years of age. Manifestations of MPS II may include not inflammatory joint stiffness, with associated restriction of movements; and coarsening of facial features, including thickening of the lips, tongue (macroglossia), and nostrils. Affected children may also have an abnormally large head (macrocephaly), a short neck and broad chest, delayed tooth eruption, progressive hearing loss, enlargement of the liver and spleen (hepatosplenomegaly), cardiac valve disease and progressive growth delays resulting in short stature. Two relatively distinct clinical forms of MPS II have been recognized. In the non-neuronopathic form (formerly defined as slowly progressive milder form) of the disease, intelligence may be normal or only slightly impaired. In the neuronopathic form of the disease (formerly called early progressive more severe form), intellectual disabilities may be apparent in the early life of the patient.
MPS II is an X-linked genetic condition that mostly affects males; although a few females have been described as well, and is caused by changes (mutations) of the IDS gene that regulates the production of the iduronate 2-sulfatase enzyme. This enzyme is needed to break-down complex sugars, known as glycosaminoglycans, produced in the body.
Introduction
MPS II is one of a group of seven hereditary metabolic diseases known as the mucopolysaccharidoses, which in turn, are part of a group known as lysosomal storage disorders. Lysosomes function as the primary digestive units within cells. Enzymes within lysosomes break down or digest particular nutrients, such as certain carbohydrates and fats. In individuals with MPS disorders, including MPS II, deficiency or improper functioning of lysosomal enzymes leads to an abnormal accumulation of certain complex carbohydrates in cells (mucopolysaccharides, also known as glycosaminoglycans) within various tissues, such as the skeleton, joints, brain, spinal cord, heart, spleen, or liver.
In the neuronopathic form of this disorder, physical and mental development reaches a peak at 2-4 years of age with subsequent deterioration. Recurrent upper respiratory infections, a chronic runny nose, hearing impairment, liver and spleen enlargement, inguinal and abdominal hernias, joint stiffness and multiplex dysplasia, compression of tendons in the wrist (carpal tunnel syndrome), and joint stiffness which can result in reduction of hand function, growth failure and valvular disease commonly occur with this form of MPS II. Coarsening of the facial features with thickening of the nostrils, lips and tongue usually occur between 2 and 4 years of age. Hydrocephalus (fluid buildup in the cavities deep within the brain) is commonly found in this form of MPS II after 4 years of age. Thick skin, short neck, widely spaced teeth, and hearing loss of varying degree are also commonly present. Nodular skin lesions on the arm or the posterior chest wall, extra- high arched feet (pes cavus) and diarrhea may also occur.
Corneal clouding and cervical spine compression (very common in other MPSs) are less frequent in MPSII thank in other MPSs.
In the non-neuronopathic form of MPS II, mental function is usually normal, while the systemic complications are similar, although sometimes a reduced severity.
The gene responsible for MPS II is known as the iduronate 2-sulfatase (IDS) gene. In many individuals with MPS II, the condition is caused be relatively small changes (e.g., certain missense or nonsense mutations) in the IDS gene, or deletion or insertion of genetic material (e.g., single-base deletions or insertions) that affects gene function. Less commonly, MPS II may result from complete absence or major structural changes of the IDS gene, linking to a neuronopathic disease. Different mutations or structural changes of the gene account for the wide variability of symptoms and findings associated with the disorder (i.e., neuronopathic and non-neuronopathic types).
The IDS gene is responsible for production of the lysosomal enzyme iduronate 2-sulfatase. The deficiency of the enzyme results in an abnormal accumulation of certain complex carbohydrates (glycosaminoglycans also known as mucopolysaccharides) within the cells of various tissues of the body.
MPS II is an X-linked recessive genetic condition. X-linked genetic disorders are conditions caused by an abnormal gene on the X chromosome inherited by the mother and manifest mostly in males. Females that have an altered gene present on one of their X chromosomes are carriers for that disorder. Carrier females usually do not display symptoms because females have two X chromosomes and only one carries the altered gene. Males have one X chromosome that is inherited from their mother and if a male inherits an X chromosome that contains an altered gene he will develop the disease.
Female carriers of an X-linked disorder have a 25% chance with each pregnancy to have a carrier daughter like themselves, a 25% chance to have a non-carrier daughter, a 25% chance to have a son affected with the disease and a 25% chance to have an unaffected son.
If a male with an X-linked disorder is able to reproduce, he will pass the altered gene to all of his daughters who will be carriers. A male cannot pass an X-linked gene to his sons because males always pass their Y chromosome instead of their X chromosome to male offspring.
MPS II typically affects only males, with symptoms becoming apparent at approximately 2-4 years of age. The disorder occurs in approximately 1 in 100,000 to 1 in 170,000 male births. A few affected females have been described, due to the selective inactivation of the X chromosome inherited by the father.
The signs and symptoms of MPS II vary greatly, so the diagnosis cannot be made by physical exam alone. Diagnosis requires documentation of reduced or absent iduronate 2-sulfatase enzyme activity in blood or skin cells. A very simple determination of glycosaminoglycans in the urine may help in screening potential positive patients. Patients with MPSII disease accumulate heparin and dermatan sulfates in the urines. Molecular genetic testing for mutations in the IDS gene is available to confirm the diagnosis. It is always important to rule out a multiple sulfatase deficiency by testing other sulfatase enzymes.
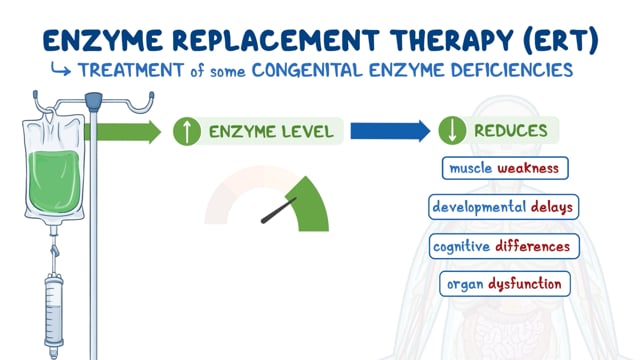
An enzyme replacement therapy, idursulfase (Elaprase), was approved in 2006 by the U.S. Food and Drug Administration (FDA) as a treatment for MPS II. This drug is sponsored by Shire.
Other treatments of MPS II are symptomatic and supportive. These management interventions commonly include developmental, occupational and physical therapy. Hernias and joint contractures may be corrected by surgery. Tonsillectomy and adenoidectomy (surgical removal of both the tonsils and the adenoids) can be used to alleviate breathing and swallowing problems. Positive pressure ventilation (CPAP or tracheostomy) can be used to apply mild air pressure to keep the airway open to alleviate breathing issues. Carpal tunnel release can treat carpal tunnel syndrome. Surgical implantation of a ventricular shunt may be used to treat possible hydrocephalus. Hearing devices may be prescribed to treat hearing loss. Developmental, physical, and occupational therapy can be helpful.
Genetic counseling is recommended for families that have a child with MPS II.
Information on current clinical trials for multiple myeloma is posted on the Internet at www.clinicaltrials.go. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government website.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Tollfree: (800) 411-1222
TTY: (800) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website:
https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, contact:
www.centerwatch.com
For information about clinical trials conducted in Europe, contact:
https://www.clinicaltrialsregister.eu/
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government website.
For information about clinical trials being conducted at the National Institutes of Health (NIH) in Bethesda, MD, contact the NIH Patient Recruitment Office:
Tollfree: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
For information about clinical trials sponsored by private sources, contact:
www.centerwatch.com
For information about clinical trials conducted in Europe, contact:
https://www.clinicaltrialsregister.eu/
TEXTBOOKS
Jones KL. Smith’s Recognizable Patterns of Human Malformation. 5th ed. Philadelphia, PA; W.B. Saunders Company; 1997:462-463.
Gorlin RJ, et al., eds. Syndromes of the Head and Neck. 3rd ed. New York, NY; Oxford University Press; 1990:106-108.
Buyse ML. Birth Defects Encyclopedia. Dover, MA; Blackwell Scientific Publications, Inc.; 1990:1162-1163.
JOURNAL ARTICLES
Coppa GV, Buzzega D, Zampini L, et al. Plasmatic and Urinary Glycosaminoglycans Characterization in Mucopolysaccharidosis II Patient Treated with Enzyme-Replacement Therapy with Idursulfase. JIMD Reports. 2012;4:79-90. doi:10.1007/8904_2011_75.
Wraith JE, Scarpa M, Beck M, et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. European Journal of Pediatrics. 2008;167(3):267-277. doi:10.1007/s00431-007-0635-4.
INTERNET
Scarpa M. Mucopolysaccharidosis Type II. 2007 Nov 6 [Updated 2018 Oct 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1274/ Accessed August 6, 2019.
Online Mendelian Inheritance in Man, OMIM). The John Hopkins University. Mucopolysaccharidosis type II. Entry Number 309900; Last Edit Date: 07/12/2019. Available at: https://omim.org/entry/309900 Accessed August 6, 2019.

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportOnline Mendelian Inheritance In Man (OMIM) has a summary of published research about this condition and includes references from the medical literature. The summary contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. OMIM is authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine.
View reportGeneReviews has an article on this condition covering diagnosis, management, and inheritance. Each article is written by one or more experts on the specific disease and is reviewed by other specialists. The article contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. The GeneReviews database is managed by the University of Washington.
View reportPlease complete this form to access the requested resource.
