

Last updated:
11/27/2023
Years published: 1987, 1988, 1990, 2004, 2006, 2009, 2012, 2015, 2018
NORD gratefully acknowledges Dag Malm, MD, PhD, Researcher, Tromso Centre of Internal Medicine (TIS as), Tromso, Norway, for assistance in the preparation of this report; and Line Borgwardt, MD, Department of Clinical Genetics, Centre for Inherited Metabolic Diseases, Copenhagen University, Copenhagen, Denmark, for critical review of the therapy section.
Summary
Alpha-mannosidosis is a rare genetic disorder characterized by a deficiency of the enzyme alpha-D-mannosidase. Alpha-mannosidosis is best thought of as a continuum of disease that is generally broken down into three forms: a mild, slowly progressive form (type 1); a moderate form (type 2); and a severe, often rapidly progressive and potentially life-threatening form (type 3). The symptoms and severity of the disorder are highly variable. Symptoms may include distinctive facial features, skeletal abnormalities, hearing loss, intellectual disability and dysfunction of the immune system. Alpha-mannosidosis is caused by changes (variants or mutations) in the MAN2B1 gene. This condition is inherited in an autosomal recessive pattern.
Introduction
Alpha-mannosidosis belongs to a group of diseases known as the lysosomal storage disorders. Lysosomes are particles bound in membranes within cells that function as the primary digestive units. Enzymes within the lysosomes break down or digest particular nutrients, such as complex molecules composed of a sugar attached to a protein (glycoproteins). Low levels or inactivity of the alpha-mannosidase enzyme leads to the abnormal accumulation of compounds upstream in the metabolic pathway in the cells of affected individuals with unwanted consequences.
The symptoms, progression and severity of alpha-mannosidosis vary widely from one person to another, including between siblings who share the same gene variant. Alpha-mannosidosis represents a spectrum or continuum of disease and is highly individualized. Some individuals develop symptoms shortly after birth and may develop potentially life-threatening complications in infancy or early childhood. Other individuals develop more moderate symptoms usually with onset before the age of 10. Some individuals may not be diagnosed until adulthood.
The disorder is generally broken down into three separate subtypes: mild (type 1), moderate (type 2) and severe (type 3). Most affected individuals fall into the moderate subtype. It is important to note, because of the highly variable nature of the disorder, that affected individuals will not have all of the symptoms discussed below.
The mild form (type 1) may not be evident until the teen years and progresses slowly. Symptoms typically include muscle weakness. Skeletal abnormalities are usually not present. The person with type 1 may have normal cognitive and physical development. However, even this later-onset form may be accompanied by mild to moderate intellectual disability. In some cases, the clinical progression of the disease appears to slow down or stop as the person grows beyond school age.
In the moderate form of the disorder (type 2), signs of skeletal abnormalities and muscle weakness may appear before ten years of age and progress slowly. Ataxia (an impaired ability to coordinate voluntary movements) may develop by the age of 20-30.
The severe form (type 3) begins within the first year of life. Most affected infants appear normal at birth, but the condition grows progressively worse. Type 3 alpha-mannosidosis is characterized by rapid progression of intellectual disability, hydrocephalus, progressive impairment of the ability to coordinate voluntary movements (ataxia), enlargement of the liver and spleen (hepatosplenomegaly), skeletal abnormalities and coarse facial features.
Intellectual disabilities associated with alpha-mannosidosis can range from mild cognitive impairment to profound mental deficiency. The severity can vary dramatically even among siblings. Children often experience delays achieving the ability to speak, and their speech stays blurred.
Motor skills may also be affected in alpha-mannosidosis. Affected children may experience delays in learning to walk and may appear clumsy. Diminished muscle tone (hypotonia) is often present.
Many individuals with alpha-mannosidosis develop moderate to severe hearing loss. Hearing loss is caused by a defect of the inner ear or the auditory nerve that prevents sound vibrations from being transmitted to the brain (With normal hearing, a portion of the inner ear converts sound vibrations to nerve impulses, which are then transmitted via the auditory nerve to the brain.). Hearing can worsen even further with ear infections due to accumulation of fluid in the middle ear.
Types 2 and 3 also may be characterized by distinctive facial features including widely spaced or unevenly developed teeth, a thickened, enlarged tongue (macroglossia), prominent forehead, flattened nasal bridge and a protruding lower jaw (prognathism). Eye abnormalities may include an inability to align the eyes (strabismus or crossed eyes), clouding (opacity) of the transparent outer covering of the eye (cornea), farsightedness (hyperopia) and less commonly, nearsightedness (myopia).
Affected children may be especially prone to dental problems such as cavities. In addition, some infants are born with an abnormally twisted ankle (ankle equinus) or hydrocephalus, a condition in which the accumulation of excessive cerebrospinal fluid (CSF) in the skull causes pressure on the tissues of the brain.
Growth rates can fluctuate with accelerated early growth but subsequent impaired growth, causing short stature. Thin arms and/or legs with stiff joints may develop. Spinal abnormalities may lead to extreme curvature in some people. Over time, affected individuals may eventually develop degenerative disease affecting multiple joints (destructive polyarthropathy).
In type 3, a diminished or abnormal immune system response can make affected individuals more susceptible to bacterial infections, particularly of the respiratory system. Infections affecting the middle ear and gastrointestinal tract are also common. Recurrent infections are more common during the first decade of life.
Some individuals with alpha-mannosidosis develop psychiatric abnormalities such as confusion, anxiety, depression or hallucinations. These symptoms may persist for days or weeks, followed by a need for excessive amounts of sleep (hypersomnia). Psychiatric symptoms or behavioral problems occur in almost half of those affected and usually develop during adolescence or early adulthood.
Alpha-mannosidosis is caused by changes (variants or mutations) in the MAN2B1 gene. The MAN2B1 gene contains instructions for producing the enzyme lysosomal alpha-mannosidase (LAMAN). This enzyme is essential for breaking down (metabolizing) certain glycoproteins. Without proper levels of functional version of this enzyme, these glycoproteins abnormally accumulate in and damage various tissues and organs of the body. Variants of the MAN2B1 gene result in the lack of production of the alpha-D-mannosidase enzyme or the production of a defective, inactive form of the enzyme.
Alpha-mannosidosis is inherited in an autosomal recessive pattern. Recessive genetic disorders occur when an individual inherits a mutated gene from each parent. If an individual receives one normal gene and one mutated gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the mutated gene and have an affected child is 25% with each pregnancy. The risk of having a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
The prevalence of alpha-mannosidosis is estimated to be 1 in every 500,000 people in the general population. Alpha-mannosidosis affects males and females in equal numbers and can potentially affect individuals of any ethnic group worldwide.
A diagnosis of alpha-mannosidosis is suspected based upon identification of characteristic findings, a thorough clinical evaluation, a detailed a patient history and specialized tests that can detect deficient levels or activity of the enzyme alpha-mannosidase in white blood cells (leukocytes) or cultured tissue cells (fibroblasts).
A diagnosis of alpha-mannosidosis can be confirmed through molecular genetic testing which can reveal the variant in the MAN2B1 gene that causes the disorder. Molecular genetic testing is available on a clinical basis.
Elevated levels of certain mannose-rich oligosaccharides (a complex carbohydrate) may be found through urinary analysis. Although this finding is considered suggestive of alpha-mannosidosis, it is not diagnostic.
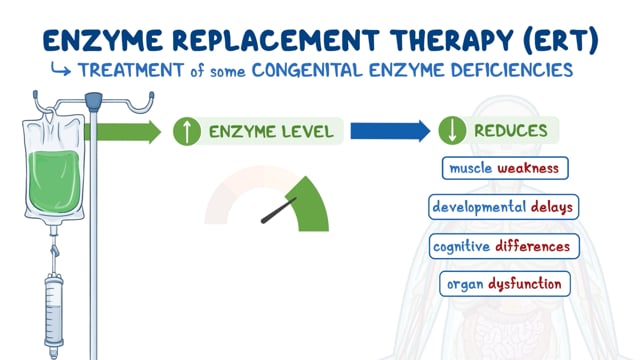
Treatment
In 2023, the U.S. Food and Drug Administration (FDA) approved velmanase alfa (Lamzede) as the first enzyme replacement therapy to treat the non-central nervous system manifestations of alpha-mannosidosis.
Other treatment for patients with alpha-mannosidosis is symptomatic and supportive. Therapy is directed at preventing and treating the complications of the disorder. Thus, it is important to be pro-active. Antibiotics are used to suppress bacterial infections. Hearing aids and pressure equalizing tubes are used to improve hearing. Physiotherapy for muscle weakness is often prescribed.
Orthopedic interventions including surgery or the use of assistive devices (e.g., special shoes or orthosis) may be necessary to treat associated skeletal abnormalities. Some individuals may require the use of a wheelchair.
Hydrocephalus may be treated by the insertion of a tube (shunt) to drain excess cerebrospinal fluid (CSF) away from the brain and into another part of the body where the CSF can be absorbed.
Early intervention is important in ensuring that children with alpha-mannosidosis reach their highest potential. Services that may be beneficial may include special education, speech therapy, special services for children with hearing loss and other medical, social and/or vocational services.
Genetic counseling is recommended for patients and their families.
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Tollfree: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website: https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, contact:
https://www.centerwatch.com/
For information about clinical trials conducted in Europe, contact:
https://www.clinicaltrialsregister.eu/
Bone marrow transplantation (BMT) is under investigation for the treatment of lysosomal storage disorders such as alpha-mannosidosis. In alpha-mannosidosis all cells are missing LAMAN. The rationale of BMT is to substitute the blood system of the patient with BMT from a donor with blood cells that can produce LAMAN. Early diagnosis and prompt treatment with a bone marrow transplant increases the chance of preventing cognitive decline and improving symptoms.
More research is necessary to determine the long-term safety and effectiveness of this potential therapy for alpha-mannosidosis. The procedure carries the risk of serious complications including graft-versus-host disease and other long-term and late effects as described by Myranek in 2011 where 2 of 17 patients died of procedure-related causes, two developed severe and six developed chronic graft-versus-host-disease (GvHD). After BMT, patients made developmental progress, although normal development was not achieved. The potential benefits of a BMT must be weighed against such drawbacks. The perfect donor is familial HLA-identical, but in many patients this donor cannot be found, in which case enzyme replacement therapy (ERT) is the best option.
Gene therapy is also being studied as another possible approach to therapy for some lysosomal storage disorders. In gene therapy, the disease-causing gene variant present in a patient is replaced with a normal gene to enable the production of active enzyme and prevent the development and progression of the disease. Given the permanent transfer of the normal gene, which is able to produce active enzyme at all sites of disease, this form of therapy is theoretically most likely to lead to a “cure.” However, at this time, there are many technical difficulties to resolve before gene therapy can succeed.
Contact for additional information about alpha-mannosidosis:
Dr. med. Dag Malm
Tromso Centre of Internal Medicine (TIS as)
Sjogata 31/33
NO9008 Tromso
Norway
Phone: +4790152552
Email: [email protected]
Website: www.tis.no
TEXTBOOKS
Malm D. Alpha-Mannosidosis. In: NORD Guide to Rare Disorders. Philadelphia, PA: Lippincott Williams & Wilkins; 2003:467-68.
Berkow R., ed. The Merck Manual-Home Edition. 2nd ed. Whitehouse Station, NJ: Merck Research Laboratories; 2003.
Leroy JG. Oligosaccharidoses and allied disorders. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR. Emory and Rimoin’s Principles and Practice of Medical Genetics, eds. 4th ed. New York, NY: Churchill Livingstone; 2002:2688-92.
Thomas GH. Disorders of glycoprotein degradation: a-mannosidosis, B-mannosidosis, fucosidosis, and sialidosis. In: Scriver CR, Beaudet AL, Sly WS, et al, eds. The Metabolic Molecular Basis of Inherited Disease. 8th ed. New York, NY: McGraw-Hill Companies; 2001:3510-15.
JOURNAL ARTICLES
Borgwardt L, Guffon N, Amraoui Y, et al.J Inherit Metab Dis. Efficacy and safety of Velmanase alfa in the treatment of patients with alpha-mannosidosis: results from the core and extension phase analysis of a phase III multicentre, double-blind, randomised, placebo-controlled trial. J Inherit Metab Dis. 2018; May 30. doi: 10.1007/s10545-018-0185-0. [Epub ahead of print]
Stroobants S, Damme M, Van der Jeugd A, et al. Long-term enzyme replacement therapy improves neurocognitive functioning and hippocampal synaptic plasticity in immune-tolerant alpha-mannosidosis mice. Neurobiol Dis. 2017; 106:255-268.
Damme D, Stroobants S, Lüdemann M, et al. Chronic enzyme replacement therapy ameliorates neuropathology in alpha-mannosidosis mice. Ann Clin Transl Neurol. 2015; 2(11):987-1001.
Beck M, Olsen JK, Wraith JR. Natural history of alpha mannosidosis a longitudinal study.Orphanet Journal of Rare Diseases 2013, 8:88.
Borgwardt L, Dali CI, Fogh J, et al. Enzyme replacement therapy for alpha-mannosidosis: 12 months follow-up of a single centre, randomised, multiple dose study.
J Inherit Metab Dis. 2013 Nov;36(6):1015-24.
Mynarek M, Tolar J, Albert MH, et al. Allogeneic hematopoetic SCT for alpha-mannosidosis: An analysis of 17 patients. Bone Marrow Transplant 2012; 47 (3):352-9.
Malm D, Pantel J, Linaker OM. Psychiatric symptoms in alpha-mannosidosis. J Intellect Disabil Res. 2005; 49:865-71.
Krivit W. Allogeneic stem cell transplantation for the treatment of lysosomal and peroxisomal metabolic diseases.Springer SeminImmunopathol. 2004; 26:119-32.
Gutschalk A, Harting I, Cantz M, et al. Adult alpha-mannosidosis: clinical progression in the absence of demyelination. Neurology. 2004; 63:1744-46.
Roces DP, Lullman-Rauch R, Peng J, et al. Efficacy of enzyme replacement therapy in alpha-mannosidosis mice: a preclinical animal study. Hum Mol Genet. 2004; 13:1979-88.
Grewal SS, Shapiro EG, Krivit W, et al. Effective treatment of alpha-mannosidosis by allogeneic hematopoietic stem cell transplantation. J Pediatr. 2004; 144:569-73.
Hansen G, Berg T, RiiseStensland HM, et al. Intracellular transport of human lysosomal alpha-mannosidase and alpha-mannosidosis-related mutants.Biochem J. 2004;381:537-46.
Peters C, Steward CG. National Marrow Donor Program, et al. Hematopoietic cell transplantations for inherited metabolic diseases: an overview of outcomes and practice guidelines. Bone Marrow Transplant. 2003; 31:229-39.
Mulrooney DA, Davies SM, Walk D, et al. Late occurrence of chronic immune-mediated axonal polyneuropathy following bone marrow transplant for juvenile-onset alpha-mannosidosis. Bone Marrow Transplant. 2003; 32:953-55.
Sun H, Wolfe JH. Recent progress in lysosomal alpha-mannosidase and its deficiency.ExpMol Med. 2001;33:1-7.
Roces DP, Lüllmann-Rauch R, Peng J, et al.: Efficacy of enzyme replacement therapy in alpha-mannosidosis mice: a preclinical animal study. Hum Mol Genet. 2004, 13: 1979-1988. 10.1093/hmg/ddh220.
Walkley SU, Thrall MA, Dobrenis K, et al. Bone marrow transplantation corrects the enzyme defect in neurons of the central nervous system in a lysosomal storage disease. Proc Natl Acad Sci USA. 1994; 91: 2970-2974. 10.1073/pnas.91.8.2970.
INTERNET
Malm D, Nilssen Ø. Alpha-Mannosidosis. 2001 Oct 11 [Updated 2012 May 3]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1396/ Accessed August 8, 2018.
Malm D, Nilssen O. Alpha-mannosidosis. Orphanet Journal of Rare Diseases. https://www.ojrd.com/content/pdf/1750-1172-3-21.pdf July 23, 2008. Accessed August 8, 2018.

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportOnline Mendelian Inheritance In Man (OMIM) has a summary of published research about this condition and includes references from the medical literature. The summary contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. OMIM is authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine.
View reportGeneReviews has an article on this condition covering diagnosis, management, and inheritance. Each article is written by one or more experts on the specific disease and is reviewed by other specialists. The article contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. The GeneReviews database is managed by the University of Washington.
View reportMedlinePlus has information about this condition that may include a description, frequency, causes, inheritance, and links to more information. The information is written for the public, including patients, caregivers and families. MedlinePlus is a service of the National Library of Medicine (NLM), which is part of the National Institutes of Health (NIH).
View reportPlease complete this form to access the requested resource.

