

Last updated:
June 06, 2019
Years published: 1986, 1994, 1995, 1997, 1998, 1999, 2001, 2002, 2003, 2004, 2007, 2009, 2012, 2015, 2019
NORD gratefully acknowledges Jaya Ganesh, MD, Associate Professor, Genetics and Genomic Medicine, Icahn School of Medicine at Mount Sinai and R.J. Desnick, PhD, MD, Dean for Genetic and Genomic Medicine, Professor and Chairman Emeritus, Icahn School of Medicine at Mount Sinai, and for assistance in the preparation of this report.
Fabry disease is a rare inherited disorder of glycosphingolipid (fat) metabolism resulting from the absent or markedly deficient activity of the lysosomal enzyme, α-galactosidase A (α-Gal A). This disorder belongs to a group of diseases known as lysosomal storage disorders. This enzymatic deficiency is caused by alterations (mutations) in the α-galactosidase A (GLA) gene that instructs cells to make the α-galactosidase A (α-Gal A) enzyme. Lysosomes function as the primary digestive tract of cells. Enzymes within lysosomes break down or digest particular compounds and intracellular structures. α-Gal A functions to break down complex sugar-lipid molecules called glycolipids, specifically, globotriaosylceramide (GL-3 or Gb3), its deacylated form Lyso-GL-3/Gb3 and related glycolipids, by removing the terminal galactose sugar from the end of these glycolipid molecules. The enzyme deficiency causes a continuous build-up of GL-3/Gb3 and related glycolipids in the body’s cells, resulting in the cell abnormalities and organ dysfunction that particularly affect small blood vessels, the heart and kidneys (Desnick 2001, Germain 2010).
The GLA gene is located on the X-chromosome and therefore, Fabry disease is inherited as an X-linked disorder. Males with the type 1 classic and type 2 later-onset phenotypes (see below) are typically significantly more severely affected than their affected female relatives (Arends 2017). Females typically have a more variable course and may be asymptomatic or as severely affected as their male relatives (see Genetics section below).
There are two major disease phenotypes: type 1 “classic” and type 2 “later-onset” subtypes. Both lead to renal failure, and/or cardiac disease, and early death (Desnick 2001, Desnick and Banikazemi 2006, Arends 2017, Doheny 2018). Type 1 males have little or no functional α-Gal A enzymatic activity (<3% of normal mean activity), and marked accumulation of GL-3/Gb3 and related glycolipids in capillaries and small blood vessels which cause the major symptoms in childhood or adolescence. These include acroparesthesias (excruciating pain in the hands and feet which occur with exercise, fevers, stress, etc.); angiokeratomas (clusters of red to blue rash-like discolorations on the skin); anhidrosis or hypohidrosis (absent or markedly decreased sweating); gastrointestinal symptoms including abdominal pain and cramping, and frequent bowel movements; and a characteristic corneal dystrophy (star-burst pattern of the cornea seen by an slit-lamp ophthalmologic examination) that does not affect vision (Sher 1979, Desnick 2001). With increasing age, the systemic GL-3/Gb3 deposition, especially in the heart leads to arrhythmias, left ventricular hypertrophy (LVH) and then hypertrophic cardiomyopathy (HCM), and in the kidneys to progressive proteinuria, renal insufficiency, and renal failure, and/or to cerebrovascular disease including transient ischemic attacks (TIAs) and strokes. Prior to renal replacement therapy (i.e., dialysis and transplantation) and enzyme replacement therapy (ERT), the average age of death of affected males with the type 1 classic phenotype was ~40 years (Columbi 1967). The incidence of males with the type 1 classic phenotype is about 1 in 40,000 (Desnick 2001), but varies with geographic region and race, ranging from about ~1 in 18,000 to 1 in 95,000 based on newborn screening studies (e.g., Spada 2006, Hwu 2009, Burlina 2018, and Wasserstein 2019).
In contrast, males with the type 2 “later-onset” phenotype (previously called cardiac or renal variants) have residual α-Gal A activity, lack GL-3/Gb3 accumulation in capillaries and small blood vessels, and do not show the early manifestations of type 1 males (i.e., the acroparesthesias, hypohidrosis, angiokeratomas, corneal dystrophy, etc). They experience an essentially normal childhood and adolescence, and typically present with renal and/or cardiac disease in the third to seventh decades of life. Most type 2 later-onset patients have been identified by enzyme screening of patients in cardiac, hemodialysis, renal transplant, and stroke clinics (Doheny 2018), and recently by newborn screening (e.g. Spada 2006, Hwu 2009, Burlina 2018, Wasserstein 2019). Based on these screening studies the incidence of type 2 later-onset disease in males varies by demography, ethnicity, and race, but is at least 5-10 times more frequent than that of the type 1 males from the same region, ethnic group, or race.
Clinical manifestations in heterozygous females from families with the type 1 classic phenotype are variable due to random X-chromosomal inactivation (Dobrovolny 2005, Echevarria 2015) and range from asymptomatic to as severe as type 1 classic males (Desnick and Banikazemi 2006, Arends 2017). Type 2 heterozygotes may be asymptomatic or develop renal or cardiac manifestations later in life. Approximately 90% of type 1 heterozygotes have the characteristic corneal dystrophy, while the type 2 heterozygous females typically lack the characteristic corneal findings or other early type 1 manifestation (Desnick 2001, Desnick and Banikazemi 2006, Doheny 2018). The frequency and severity of manifestations in type 2 heterozygous females has only been systematically investigated recently, and they are typically less frequent and less severe than those seen in their type 2 male relatives (Arends 2017).
Type 1 Classic Phenotype
The signs and symptoms of males with the type 1 classic phenotype typically begin in childhood or adolescence (Desnick 2001, Desnick and Brady 2004). Symptoms increase with age primarily due to the progressive glycolipid accumulation in the micro-vascular system, kidney podocytes, and cardiomyocytes leading to kidney insufficiency and failure, heart disease, and/or strokes. Early and progressive clinical symptoms include:
Common Manifestations in Type 1 and 2 Males
With advancing age in type 1 males, typically in the third to fourth decades, and in type 2 males in the third to sixth decades, the progressive GL-3/Gb3 glycolipid deposition leads to renal and/or heart manifestations as described below (Desnick 2001, Arends 2017). Many of the type 2 later-onset males who lack the early manifestations seen in the type 1 males, are detected in renal, heart, or stroke clinics (Nakao 1995, 2003; Doheny 2018). Patients with the type 2 later-onset subtype typically do not have the skin lesions (angiokeratoma), sweat normally, do not experience the Fabry pain or crises, and do not have heat intolerance or corneal involvement. These individuals develop heart or kidney disease later in adult life.
Signs of progressive organ involvement include:
Renal dysfunction. Progressive decrease in renal function is due to the progressive accumulation of GL-3/Gb3 in the kidneys, particularly in the endothelial cells, smooth muscle cells and podocytes (Najafian 2013; Tondel 2008, 2013). There is histological evidence of this accumulation and ensuing cellular and vascular injury to renal tissue beginning in childhood and adolescence (Tondel 2008, 2013; Najafian 2013) in type 1 classic males and females. In type 1 classic males, the decline in typically begins with podocyte involvement and microalbuminuria leading to frank proteinuria, increasing loss of function (decreasing glomerular filtration rate or GFR), all leading to kidney failure and the need for dialysis or transplantation typically by 35 to 45 years of age. In type 2 males, kidney involvement typically occurs in the fourth decade or later, but some patients do not develop renal failure (Meehan 2004). Kidney involvement in type 1 female heterozygotes is more variable. Only about 10-15% of type 1 females develop kidney failure. It is not clear what percentage of type 2 females develop renal failure, if any (Arends 2017).
Cardiac disease. GL-3/Gb3 deposition can be found in all cardiac tissues, including valves, cardiomyocytes, nerves, and coronary arteries (Desnick 1976). Heart disease includes heart enlargement, typically left ventricular hypertrophy (LVH) leading to hypertrophic cardiomyopathy (HCM), rhythm abnormalities (arrhythmias), and heart failure (Frustaci 2017). LVH occurs in about 20% of males and females with an average age of diagnosis in the early 20s to 40s among type 1 males and late 30s to 40s among type 1 female heterozygotes. Early heart involvement in type 1 males typically includes arrhythmias and mitral insufficiency in their 20s followed by LVH leading to HCM. Type 2 later-onset males develop similar heart manifestations as type 1 males, but at older ages and may be first diagnosed in cardiac clinics among patients with LVH or HCM (Doheny 2018). Heterozygous females with the type 1 phenotype often have sinus bradycardia as an early finding and may more severely affected heterozygotes can develop LVH progressing to HCM.
Cerebrovascular complications. As a result of the progressive GL-3/Gb3 deposition in the heart leading to atrial fibrillation and in the small blood vessels in the brain, about 7% of males and 4% of females with Fabry disease, particularly those with the type 1 phenotype, experience ischemic or hemorrhagic strokes, occurring typically in the fourth decade of life or later (Fellgiebel 2006, Wilcox 2008).
Respiratory abnormalities: Accumulation of glycosphingolipids and consequent fibrosis can cause interstitial lung disease. Pathological changes and tissue remodeling may involve both alveoli and the bronchial tree leading to restrictive lung disease, obstructive airway disease, or a mixture of obstructive and restrictive disease. (Svensson 2015). Respiratory symptoms may occur independent of cardiovascular disease in these patients.
Other pathology: Hearing loss, tinnitus, dizziness and vertigo potentially due to GL-3/Gb3 deposition in vestibular structures and/or auditory neuropathy are commonly reported in adult patients and while these are not life threatening, contribute to disease burden and negatively affect quality of life. Depression has been reported and a portion of these cases, especially Type I Classic males, were classified as having severe depression. (Cole 2007)
Genetics
Fabry disease is caused by alterations (mutations) in the alpha-galactosidase A (GLA) gene located on the X-chromosome. Chromosomes are found in the nucleus of all cells. They carry the genetic characteristics of each individual in thousands of specific segments, called “genes” that span the length of the chromosomes. Each of these genes has a specific function in the body. Human chromosomes are organized in pairs, numbered from 1 through 22, with the 23rd pair of X- and Y-chromosomes for males and two X-chromosomes for females. Individuals inherit one chromosome in each pair from each parent. Therefore, in X-linked disorders including Fabry disease, disease traits on the X-chromosome can be masked or reduced in females by the normal gene on the other X-chromosome. More specifically, because only one functioning X-chromosome is required in males and females, one of the X-chromosomes in each cell of a female is essentially “turned off”, usually in a random pattern (random X-chromosome inactivation). This means that in X-linked disorders, some cells will have the X-chromosome with the mutated “Fabry” gene activated, while others will have the X-chromosome with the functioning, normal gene activated. Therefore, in Fabry disease the symptoms and severity of organ involvement are dependent on the percentage of cells in the tissue/organ where the X-chromosome with the GLA gene mutation is active, but with no or markedly decreased function, which partially explains why the disease severity in females is more variable than in their affected male relatives. Since males have only one X-chromosome, if a male has the X-chromosome with the GLA gene mutation, he will be affected with the disorder. Therefore, type 1 classic and type 2 later-onset males with Fabry disease are more uniformly affected, whereas symptoms in females, due to random X-inactivation, may range from asymptomatic or as severely affected as their affected male relatives (Dobrovolny 2005, Echevarria 2016)
Males with X-linked Fabry disease transmit the GLA gene mutation to all their daughters, who are heterozygotes, but never to their sons. Female heterozygotes have a 50 percent risk of transmitting the disease to each of their children, both daughters and sons, with each pregnancy.
The GLA gene normally instructs the body’s cells to make the α-Gal A enzyme, which breaks down the accumulating glycolipids (GL-3/Gb3) in the cell’s lysosomes. Fabry disease is caused by mutations in the GLA gene. There are over 965 reported mutations in the GLA gene that are responsible for Fabry disease (Stenson 2017; Human Gene Mutation Database; https://www.hgmd.org), causing the type 1 or 2 phenotypes. Two databases provide phenotype assignments for all reported mutations: dbFGP.org and Fabry-Database.org (Saito 2011). Thus, the severity and range of symptoms may vary among individuals depending on the GLA mutation in their family. Some mutations markedly alter the enzyme such that it has little to no activity. These mutations cause the type 1 classic subtype (e.g., Eng 1997, Shabber 2006), while other mutations result in a small amount of residual enzyme activity and the type 2 later-onset subtype (e.g., von Scheidt 1991, Eng 1997, Nakao 2003, Spada 2006). The signs and symptoms of Fabry disease develop due to absent or markedly deficient α-Gal A enzymatic activity. Patients with the type 1 classic phenotype, who have no or very low activity levels (less than 3% of normal), accumulate the GL-3/Gb-3 glycolipid substance (and related glycolipids) in most tissues of the body, especially small blood vessels, and certain cells in the heart and kidneys. Patients with the type 2 later-onset phenotype have residual enzyme activity (3-15% of mean normal activity, Desnick 2001), also accumulate GL-3/Gb3, but to a lesser extent and at a slower rate. They tend to have a somewhat less severe form of the disease, but males with the type 2 subtype ultimately develop severe cardiac disease and/or renal failure. There are also mutations in the GLA gene that are benign and do not cause Fabry disease (e.g., Froissart 2003, Doheny 2018)
Fabry disease is a rare pan-ethnic disorder, meaning that it occurs in all racial and ethnic populations affecting males and females. It is estimated that type 1 classic Fabry disease affects approximately one in 40,000 males. The type 2 later-onset phenotype is more frequent, than the type 1 phenotype by 3-10 fold, and in some populations may occur as frequently as about 1 in 1,500 to 4,000 males (Spada 2006, Hwu 2009, Chien 2012). Data emerging from the newborn screening studies suggests that the incidence of Fabry disease varies in different geographic regions (Spada 2006, Hwu 2009, Burlina 2018, Wasserstein 2019). Already, newborn screening for Fabry disease has been initiated in several states in the USA.
The clinical diagnosis of the type 1 classic phenotype can be made clinically by physicians who recognize the characteristic findings of episodic pain in the extremities, absent or decreased sweating (anhidrosis or hypohidrosis), typical skin lesions (angiokeratoma), gastrointestinal abnormalities, and the corneal dystrophy in childhood or adolescence (Desnick 2003). The disease progresses to renal insufficiency, and/or heart and cerebrovascular disease in adulthood. In type 2 males, the diagnosis is often missed, and may be made in adulthood when the cardiac and/or kidney disease becomes manifest. Many males with the type 2 later-onset phenotype have been diagnosed by screening patients in hemodialysis, cardiac, and stroke clinics (Doheny 2018). The diagnosis of both type 1 and 2 males is confirmed by demonstrating the enzyme deficiency and by identifying the specific GLA gene mutation.
Female heterozygotes can have α-GAL A enzymatic activity from markedly decreased to values in the normal range. Therefore, heterozygous females are only accurately diagnosed by demonstrating the specific α-galactosidase A (GLA) gene mutation.
Early prenatal diagnosis at about 10 weeks of pregnancy can be made by α-Gal A enzyme and GLA mutation analyses of villi obtained by chronic villus sampling, or by amniocentesis at about 15 weeks of gestation by determining the α-Gal A enzyme activity and demonstrating the family-specific GLA mutation (Desnick 2007). Preimplantation genetic diagnosis is available when the familial mutation in the GLA gene is known.
Newborn screening studies have identified affected males by demonstrating the reduced α-Gal A activity in dried blood spots followed by GLA gene sequencing (e.g., Spada 2006, Burlina 2018, Wasserstein 2019).
Treatment
Fabry disease causes multi-organ dysfunction and patients need a comprehensive, multi-disciplinary treatment plan that is individually tailored and includes specific therapies that target abnormal substrate accumulation and adjuvant therapies that address end-organ damage (Ortiz 2018).
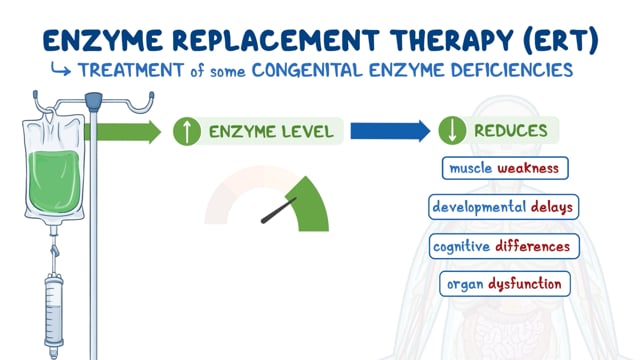
Enzyme replacement therapy (ERT) is the cornerstone for treatment of Fabry disease and synthetic enzyme, produced by recombinant DNA technology, is infused intravenously. Two forms of the recombinant enzyme are available, agalsidase alpha (Replagal®, Shire pharmaceuticals) and agalsidase beta (Fabrazyme®, Sanofi Genzyme). Fabrazyme is the only ERT approved by the Food and Drug Administration (FDA) in 2003. Both Replagal and Fabrazyme are available in Europe and other regions of the world. ERT replaces the missing enzyme and reduces the accumulated glycolipids in cells throughout the body. Double-blind, placebo-controlled Phase 3 and 4 clinical trials have demonstrated the safety and effectiveness of Fabrazyme (Eng 2001A, 2001B; Banikazemi 2007, Fellgiebel 2014, Germain 2015).
ERT has been shown to slow or prevent the decline of renal function especially if initiated early before advanced kidney damage, improve neuropathic pain and heat intolerance (Eng 2001, Germain 2015). Globotriaosylceramide accumulation is cleared from various cell types in the kidney following ERT (Tondel 2013, Skrunnes 2017). Early initiation of ERT is important especially in type 1 classically affected males. ERT initiation is currently recommended for type 1 Classic males with clnical manifestations at any age, or if asymptomatic, by age 15 (Hopkin 2016, Ortiz 2018). Recombinant enzyme “biosimilars” are available in certain countries including Korea and Japan. Several other recombinant enzyme preparations are in clinical development.
An oral therapy, Galafold (migalastat, Amicus Therapeutics) was approved in the EU (2017) and in the US (2018) to treat adults with Fabry disease. The drug is a pharmacologic chaperone that can bind to, stabilize, and enhance the residual enzymatic activity of certain missense mutations (Desnick and Schuchman 2002, Benjamin 2017). Clinical studies have demonstrated the effectiveness of this approach (Germain 2016). Future studies will determine the clinical and biochemical effectiveness of specific missense mutations with residual activity.
Adjunct therapies include low daily doses of diphenylhydantoin, carbamazepine, or neurontin, to help to manage the acroparesthesia (Burlina 2011). Other later complications (e.g., kidney failure or heart problems) should be treated symptomatically after consultation with a physician who is experienced in the care of patients with Fabry disease. Hemodialysis and kidney (renal) transplantation may be necessary in cases that have progressed to kidney failure (Thadhani 2002, Ersözlü 2018).
Genetic counseling is recommended for affected individuals and their families.
Trials of gene therapy are also underway.
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Toll-free: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website:
https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, in the main, contact:
www.centerwatch.com
For more information about clinical trials conducted in Europe, contact:
https://www.clinicaltrialsregister.eu/
Contact for additional information about Fabry disease:
International Center for Fabry Disease
Icahn School of Medicine at Mount Sinai
Fifth Avenue at 100th Street
New York, NY 10029
(212) 659-6700
Toll-free: 1-866-FABRY-MD
TEXTBOOKS
Desnick R, Ioannou Y, Eng C. Alpha-Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease.8th ed. New York, NY: McGraw-Hill; 2001:3733-3774.
JOURNAL ARTICLES
Arends, M, et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J Am Soc Nephrol. 2017; 28:1631-1641.
Balwani M, Fuerstman L, Kornreich R, Edelmann L, Desnick RJ. Type I Gaucher disease: Significant disease manifestations in “asymptomatic” homozygotes. Arch Intern Med. 2010; 170:1463-1469.
Banikazemi M, et al. Agalsidase-beta therapy for advanced Fabry disease: A randomized trial. Ann Intern Med. 2007; 146:77-86.
Benjamin ER, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. 2017; 19:430-443.
Burlina AB, Polo G, Salviati L, Duro G, Zizzo C, Dardis A, etc. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J Inherit Metab Dis. 2018; 41:209-219.
Burlina AP, Sims KB, Politei JM, Bennett GJ, Baron R, Sommer C, Møller AT, Hilz MJ. Early diagnosis of peripheral nervous system involvement in Fabry disease and treatment of neuropathic pain: the report of an expert panel. BMC Neurol. 2011; 11:61.
Cole Al, Lee PJ, Hughes D, Deegan PB, Waldek S, Lachman RH. Depression in adults with Fabry disease: A common and under-diagnosed problem. J Inherit Metab Dis. 2007; 30:943–951
Chien YH, Ni-Chun L, Hian SC, Desnick RJ, Hwu WL: Fabry disease: Incidence of the common later-onset α-galactosidase A IVS4+919G>A mutation in Taiwanese newborns – Superiority of DNA-based to enzyme-based newborn screening for common mutations. Mol Med. 2012; 18:780-784.
Columbi A, Kostyal A, Bracher R, Gloor F, Mazzi R, Thölen H. Angiokeratoma corporis diffusum – fabry Disease. Helv Med Acta. 1967; 34:67-83
Desnick RJ, Schuchman EH. Enzyme replacement and enhancement therapies: Lessons from lysosomal disorders. Nature Rev Genet. 2002; 3:954-966.
Desnick RJ, et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003; 138:338-346.
Desnick RJ, Brady RO. Fabry disease in childhood. J. Pediatr. 2004; 144:520-526.
Desnick RJ and Banikazemi M. Fabry disease: Clinical spectrum and evidence-based enzyme replacement therapy. Nephrol Ther. 2006; Suppl 2:S172-S185.
Desnick RJ. Prenatal diagnosis of Fabry disease. Prenat Diag. 2007; 27:693-694.
Dobrovolny R, et al. Relationship between X-inactivation and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the alpha-galactosidase A gene in the Czech and Slovak population. J Mol Med (Berl). 2005; 83:647-54.
Doheny D, Srinivasan R, Pagant S, Chen B, Yasuda M, Desnick RJ.: Fabry disease: Prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995-2017. J. Med Genet. 2018; 55:261-268.
Echevarria L, et al. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. 2016; 89:44-54.
Eng CM, Ashley GA, Burgert TS, Enriquez AL, D’Souza M, Desnick RJ. Fabry disease: Thirty-five mutations in the α-galactosidase A gene in patients with classic and variant phenotypes. Mol Med. 1997; 3:174-182.
Eng CM, et al. A phase 1/2 clinical trial of enzyme replacement in Fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet. 2001A; 68:711-722.
Eng CM, et al. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 2001B; 345:9-16.
Ersözlü S, et al. Long-term outcomes of kidney transplantation in Fabry disease. Transplantation. 2018; 102:1924-1933.
Fellgiebel A, et al. CNS manifestations of Fabry’s disease. Lancet Neurol. 2006; 5:791-795.
Fellgiebel A, Gartenschläger M, Wildberger K, Scheurich R, Desnick RJ, Sims K: Enzyme replacement therapy stabilized white matter lesion progression with Fabry disease. Cerebrovasc Dis. 2014; 38:448-456.
Froissart R, Guffon N, Vanier MT, Desnick RJ, Maire I. Fabry disease: D313Y is an alpha-galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol Genet Metab. 2003; 80:307-314.
Frustaci A, Chimenti C, Doheny D, Desnick, RJ: Evolution of cardiac pathology in classic Fabry disease: Progressive cardiomyocyte enlargement leads to increased cell death and fibrosis, and correlates with severity of ventricular hypertrophy. Int J Cardiol. 2017; 248:257-262.
Germain DP, et al. Fabry disease. Orphanet J Rare Dis. 2010; 5:30.
Germain DP, et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet. 2015; 52:353-358.
Germain DP, et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N Engl J Med. 2016; 375:545-555.
Hopkin RJ, Jefferies JL, Laney DA, Lawson VH, Mauer M, Taylor MR, Wilcox WR; Fabry Pediatric Expert Panel. The management and treatment of children with Fabry disease: A United States-based perspective. Mol Genet Metab. 2016; 117:104-113.
Hwu WL, et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the Later-Onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum Mutat. 2009; 30:1397-1405.
Kanzaki T, Yokota M, Irie F, Hirabayashi Y, Wang AM, Desnick RJ. Angiokeratoma corporis diffusum with glycopeptiduria due to deficient lysosomal α-N-acetylgalactosaminidase activity: Clinical, morphologic and biochemical studies. Arch Dermatol. 1993; 129:460-465.
Meehan S, Jusanto T, Rydel JJ, Desnick RJ. Fabry disease: Renal involvement limited to podocyte pathology and proteinuria in a septuagenarian cardiac variant. Pathologic and therapeutic implications. Am J Kidney Dis. 2004; 43:164-171.
Najafian B, Mauer M, Hopkin RJ, Svarstad E. Renal complications of Fabry disease in children. Pediatr Nephrol. 2013; 28:679-687.
Nakao S, et al. An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N Engl J Med. 1995; 333:288-293.
Nakao S, et al. Fabry disease: Detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003;64:801-807.
Ortiz A, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018; 123:416–427.
Saito S, Ohno K, Sakuraba H. Fabry-database.org: database of the clinical phenotypes, genotypes and mutant α-a-galactosidase A structures in Fabry disease. J Hum Genet. 2011; 56:467-468.
Schiffmann R, et al. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009; 24:2102-2111.
Schindler D, et al. Neuroaxonal dystrophy due to lysosomal α-N-acetylgalactosaminidase deficiency. Clinical, pathologic and biochemical delineation. N Engl J Med. 1989; 320:1735-1740.
Shabbeer J, Yasuda M, Benson SD, Desnick RJ. Fabry disease: identification of 50 novel alpha-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations. Hum Genomics. 2006; 2:297-309.
Sher NA, Letson RD, Desnick RJ. The ocular manifestations in Fabry’s disease. Arch Ophthalmol. 1979; 97:671-676.
Skrunes R, et al. Reaccumulation of globotriaosylceramide in podocytes after agalsidase dose reduction in young Fabry patients. Nephrol Dial Transplant. 2017; 32:807-813.
Spada M, et al. High incidence of Later-Onset Fabry disease revealed by newborn screening. Am J Hum Genet. 2006; 79:31-40.
Stenson, PD, et al. The Human Gene Mutation Database: Towards a Comprehensive Repository of Inherited Mutation Data for Medical Research, Genetic Diagnosis and Next-Generation Sequencing Studies. Hum Genet. 2017; 136:665-77.
Svensson, CK, Feldt-Rasmussen U, Backer V. Fabry disease, respiratory symptoms, and airway limitation – a systematic review. Eur Clin Respir J. 2015; 2:1-8.
Thadhani R, et al. Patients with Fabry disease on dialysis in the United States. Kidney Int. 2002; 61:249-255.
Tøndel C, Bostad L, Hirth A, Svarstad E. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis. 2008; 51:767-776.
Tøndel C, Bostad L, Larsen KK, Hirth A, Vikse BE, Houge G, Svarstad E.: Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol. 2013; 24:137-148.
Wasserstein MP, et al. The New York pilot newborn screening program for lysosomal storage diseases: Report of the First 65,000 Infants. Genet Med. 2019; 21:631-640
Wilcox WR, et al. Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry Registry. Mol Genet Metab. 2008; 93:112-128.
von Scheidt W, et al. An atypical variant of Fabry’s disease with manifestations confined to the myocardium. N Engl J Med. 1991; 324:395-399.

NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportOnline Mendelian Inheritance In Man (OMIM) has a summary of published research about this condition and includes references from the medical literature. The summary contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. OMIM is authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine.
View reportGeneReviews has an article on this condition covering diagnosis, management, and inheritance. Each article is written by one or more experts on the specific disease and is reviewed by other specialists. The article contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. The GeneReviews database is managed by the University of Washington.
View reportMedlinePlus has information about this condition that may include a description, frequency, causes, inheritance, and links to more information. The information is written for the public, including patients, caregivers and families. MedlinePlus is a service of the National Library of Medicine (NLM), which is part of the National Institutes of Health (NIH).
View reportPlease complete this form to access the requested resource.

