

Last updated:
7/23/2024
Years published: 1984, 1985, 1987, 1990, 1992, 1997, 1999, 2007, 2008, 2011, 2017, 2024
NORD gratefully acknowledges Anjali Chauhan, Medical Affairs Program Manager and Gioconda Alyea, MD (FMG), MS, National Organization for Rare Disorders, for assistance in the preparation of this report.
Summary
Cri du chat syndrome (CdCS or 5p-) is a rare genetic disorder caused by a loss (deletion) in chromosome 5. Symptoms can be different from person to person depending on the exact size and location of the deleted genetic material. Common symptoms include a unique cry that sounds like the mewing of a cat, characteristic facial features, slow growth and a head that is smaller than expected (microcephaly). Affected children also show moderate to severe intellectual disability, as well as delays in gaining skills (developmental delays) and additional symptoms. In most patients, CdCS is caused by a random (de novo) genetic error that occurs very early when the baby is developing inside the womb. Treatment is based on improving the symptoms and may include early interventional services, therapies and surgeries.
Introduction
CdCS was first described in medical literature in 1963 by Dr. Lejeune who named the disorder after the distinctive cat-like cry. In French, cri du chat translates into “cry of the cat”.
The most recognizable sign is a characteristic high-pitched, shrill cry that is present during the first few weeks of life. The cry, which sounds like the mewing of a cat, becomes less noticeable as affected infants grow older.
Signs and symptoms can be different depending on the size and location of the chromosome 5 deletion. The following signs and symptoms have been described in people affected with cri du chat syndrome:
As affected children get older, the following signs and symptoms may change:
In addition, the children may develop some developmental and behavioral problems such as:
Cri du chat syndrome is a chromosomal disorder caused by a partial loss (deletion) of part of the short arm (p) of chromosome 5. Chromosomes carry the genetic information, or genes, of all living things. Pairs of human chromosomes are numbered from 1 to 23 and each chromosome is divided into two sections (arms) based on the location of a narrowing (constriction) called the centromere. By convention, the shorter arm is known as “p”, and the larger arm is known as “q”. Cri du chat is also called 5p- because there is missing genetic information in the p arm of chromosome 5.
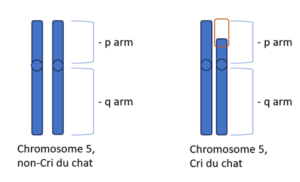
The specific symptoms and severity of symptoms depends on the size and location of the missing part of chromosome 5p. Researchers have determined that certain symptoms may be associated with specific regions on the short arm of chromosome 5. In those regions, they have identified several genes that are thought to play a role in the development of cri du chat syndrome. Some examples include the TERT gene and the SEMA3F gene. When deleted, this could contribute to the wide range of possible features that people with cri du chat have. Another example is the CTNND2 gene. Deletion of this gene is linked to more severe intellectual disability. If researchers can link specific sets of symptoms and findings (phenotypes) to specific deletions in chromosome 5p, it may help in the diagnosis and management of the condition.
Most cases of cri du chat syndrome appear to occur randomly (de novo) for unknown reasons very early in embryonic development and are not inherited from the parents. Most deletions (80-90%) are paternal in origin, meaning they likely occur as part of sperm formation in the child’s father. The parents of a child with a de novo deletion usually have normal chromosomes and a relatively low chance of having another child with this condition.
In approximately 10-15% of patients, cri du chat syndrome is caused by a balanced translocation involving chromosome 5p and another chromosome. In a balanced translocation, pieces of genetic material on two different chromosomes swap places with each other (translocate) for unknown reasons, but no material is lost or gained (balanced). Therefore, a person who has a balanced translocation does not have any problems because they do not have any gain or loss of genetic material. However, if a child inherits a chromosome 5 that was involved in a balanced translocation, the child now has a chromosome 5 that is missing genetic material, which can lead to developing cri du chat syndrome. In addition, because a parent can pass on that chromosome 5, there is a higher chance of having another child with this condition. Chromosomal analysis may determine whether a parent has a balanced translocation.
The frequency of cri du chat syndrome ranges from 1/15,000 to 1/50,000 live births. This syndrome affects females more often than males. Some people with cri du chat syndrome may go undiagnosed making it difficult to determine the true frequency of this disorder in the general population.
Cri du chat syndrome can be diagnosed before or after birth. Before birth (prenatally), the condition can be diagnosed through a procedure called amniocentesis. During amniocentesis, a sample of the fluid that surrounds the fetus in the womb (amniotic fluid), is taken for testing. The fluid contains cells from the fetus, which hold the chromosomes of the fetus and can be tested for the deletion in chromosome 5. This testing can be paired with ultrasound findings. An ultrasound may reveal physical features of the fetus that raise a suspicion of a genetic condition like cri du chat syndrome.
In newborns, the diagnosis of cri du chat syndrome may be suspected based on a thorough clinical evaluation, identification of characteristic signs and symptoms (e.g., cat-like cry). The diagnosis is confirmed with chromosomal studies (karyotyping) that reveal a deletion on the short arm of chromosome 5. Other tests that can be used to confirm a diagnosis of cri du chat syndrome are fluorescence in situ hybridization (FISH), comparative genomic hybridization (CGH), or a quantitative polymerase chain reaction (PCR).
Chromosomal studies may also be performed on the parents to determine if one of them has a balanced translocation.
Clinical Testing and Work-Up
Additional tests may be used to determine the extent of the disorder such as X-rays to look for skeletal differences such as scoliosis.
Treatment
Cri du chat syndrome is treated by managing the specific symptoms that someone has. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, orthopedists, surgeons, cardiologists, speech pathologists, neurologists, dentists, physical and occupational therapists and other health care professionals may need to systematically and comprehensively plan an affected child’s treatment.
As some children with cri du chat syndrome can have sensory-neural deafness, auditory testing should be performed.
Early intervention is important in making sure that children with cri du chat syndrome reach their highest potential. Services that may be beneficial include special remedial education, physical therapy, speech therapy, special services and other medical, social and/or vocational services. Most children are enrolled in therapies before one year of age.
Surgery may be performed to treat a variety of symptoms potentially associated with cri du chat syndrome including congenital heart defects, strabismus, scoliosis, clubfoot, cleft palate and cleft lip.
The survival for children with cri du chat syndrome is generally good. Most syndrome related deaths occur within the first year of life. Several affected individuals have lived to be over 50 years of age.
Genetic counseling is recommended for affected individuals and their families. Other treatments are symptomatic and supportive.
Research and studies of cri du chat syndrome are ongoing. One study has shown that early special schooling, a home environment (rather than an institutional one) and family support may help the patient achieve the abilities of a five- or six-year-old. In the same study, half the children over ten who had undergone special schooling and lived in a supportive home environment were able to communicate adequately.
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Tollfree: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website:
https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, contact:
www.centerwatch.com
For information about clinical trials conducted in Europe, contact:
https://www.clinicaltrialsregister.eu/
TEXTBOOKS
Mezoff AG. Cri-du-Chat Syndrome. NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia, PA. 2003:175.
JOURNAL ARTICLES
Ullah I, Mahajan L, Magnuson D. A newly recognized association of Hirschsprung disease With Cri-du-chat syndrome. Am J Gastroenterol. 2017;112:185-186.
Nguyen JM, Qualmann KJ, Okashah R, Reilly A, Alexeyev MF, Campbell DJ. 5p deletions: Current knowledge and future directions. Am J Med Genet C Semin Med Genet. 2015 Sep;169(3):224-38.
Rodriguez-Caballero A, Torres-Lagares D, Rodriguez-Perez A, Serrera-Figallo MA, Hernandez-Guisado JM, Machuca-Portillo G. Cri du chat syndrome: A critical review. Oral Patol Oral Cir Bucal. 2010;15:e473-8.
Cerruti Mainardi, P. Cri du Chat syndrome. Orphanet J Rare Dis. 2006;1: 33. https://doi.org/10.1186/1750-1172-1-33
Hill C, Moller JH, Finkelstein M, Lohr J, Schimmenti L. Cri du chat syndrome and congenital heart disease: a review of previously reported cases and presentation of an additional 21 cases from the pediatric cardiac care consortium. Pediatrics. 2006;117:924-7.
Laczmanska I, Stembalska A, Gil J, Czemarmazowicz H, Sasiadek M. Cri du chat syndrome determined by the 5p15.3→pter deletion–diagnostic problems. Eur J Med Genet. 2006;49:87-92.
Mainardi PC, Pastore G, Castronovo C, et al., The natural history of cri du chat syndrome. A report from the Italian Register. Eur J Med Genet. 2006;49:363-83.-9.
Kondoh T, ShimoKawa O, Harada N, Doi T, et al., Genotype-phenotype correlation of 5p- syndrome: pitfall of diagnosis. J Hum Genet. 2005;50:26.
Posmyk R, Panasiuk B, Yatsenko SA, Stankiewicz P, Midro AT. A natural history of a child with monosomy 5- syndrome (cat-cry/cri-du-chat syndrome) during the 18 years of follow-up. Genet Couns. 2005;16:17-25.
Zhang X, Snijders A, Segraves R, et al., High-resolution mapping of genotype-phenotype relationships in cri du chat syndrome using array comparative genomic hybridization. Am J Hum Genet. 2005;76:312-6.
Van Buggenhout GJ, Pijkels E, Holvoet M, et al., Cri du chat syndrome: changing phenotype in older patients. Am J Med Genet. 2000;90:203-15.
INTERNET
Ajitkumar A, Jamil RT, Mathai JK. Cri Du Chat Syndrome. [Updated 2022 Oct 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482460/ Accessed July 10, 2024.
Cri-du-chat syndrome: Medlineplus genetics. MedlinePlus. October 25, 2022. https://medlineplus.gov/genetics/condition/cri-du-chat-syndrome/ Accessed October 3, 2023.
McKusick VA., ed. Online Mendelian Inheritance in Man (OMIM). Baltimore. MD: The Johns Hopkins University; Entry No:123450; Last Update: 1/13/2023. Available at: http://omim.org/entry/123450 Accessed July 22, 2024.
Lal MK. Cri-du-Chat Syndrome. Updated: Mar 01, 2024. Medscape. Available at: http://www.emedicine.com/ped/topic504.htm Accessed July 8, 2024.

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportOnline Mendelian Inheritance In Man (OMIM) has a summary of published research about this condition and includes references from the medical literature. The summary contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. OMIM is authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine.
View reportMedlinePlus has information about this condition that may include a description, frequency, causes, inheritance, and links to more information. The information is written for the public, including patients, caregivers and families. MedlinePlus is a service of the National Library of Medicine (NLM), which is part of the National Institutes of Health (NIH).
View reportPlease complete this form to access the requested resource.

