

Last updated:
4/16/2025
Years published: 1990, 1991, 1992, 1994, 1996, 1997, 1998, 2000, 2001, 2004, 2005, 2022, 2025
NORD gratefully acknowledges Karl E. Anderson, MD, FACP, Galveston Porphyria Laboratory & Center, University of Texas Medical Branch/UTMB Health, for assistance in the preparation of this report.
Summary
Porphyrias are rare disorders resulting from impairment of one or more of the eight enzymes that act in sequence in the pathway to make heme. Heme is a component of important proteins known as hemoproteins. The largest amounts of heme are made in the bone marrow and liver. Therefore, in the various porphyrias, intermediate chemicals in this pathway first accumulate in the bone marrow (in erythropoietic porphyrias) or liver (in hepatic porphyrias). Erythropoietic porphyrias often first cause symptoms early in life, whereas hepatic porphyrias mostly affect adults. Intermediates chemicals late in the pathway to make heme are porphyrins, and these can be activated by light and cause skin photosensitivity, whereas those early in the pathway are called porphyrin precursors, and their accumulation is associated with effects on the nervous system.
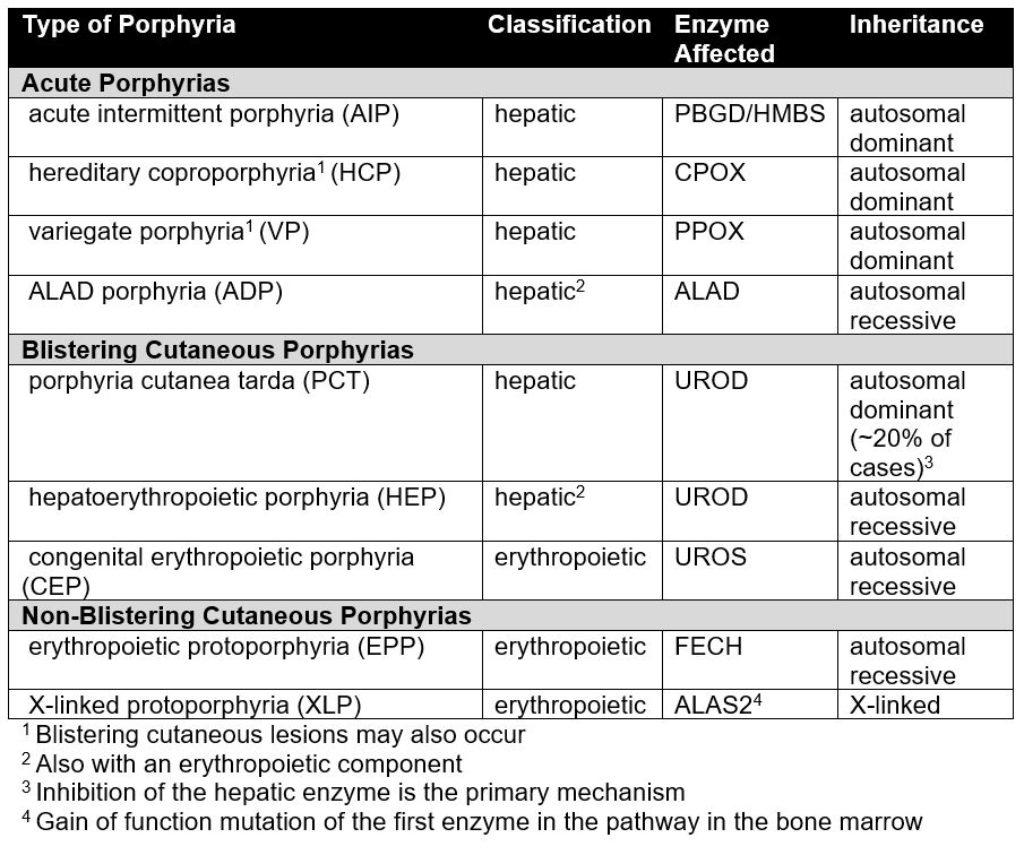
Three clinical types of porphyria are readily distinguished but are not completely distinct (see table below). The acute porphyrias, also known as the acute hepatic porphyrias, mainly affect the nervous system. Symptoms include periodic attacks of abdominal pain, other gastrointestinal symptoms, mental changes and pain and weakness of the extremities. Blistering cutaneous porphyrias are chronic and present with blistering, scarring and pigment changes of areas of skin exposed to sunlight. Non-blistering cutaneous porphyrias cause much more acute, severe and painful reactions after sunlight exposure, which greatly alters behavior but cause few blisters or chronic skin changes.
All but one of the porphyrias are related to changes (pathogenic variants) in genes for enzymes in the pathway to make heme, and these variants are inherited in families. However, many individuals with a familial variant never have symptoms. Other external and genetic factors are necessary before symptoms occur, especially in hepatic porphyrias. These may include certain medications, hormones, chemicals, infections, alcohol use or changes in diet. Non-inherited causative factors predominate in the most common porphyria, porphyria cutanea tarda, and most cases of this porphyria occur in the absence of a familial gene variant.
Diagnosis of porphyrias can be challenging because they are uncommon, and their symptoms resemble those of other more common conditions. Diagnosis requires laboratory measurements of intermediates in the pathway to make heme that are present in large amounts in red blood cells, plasma, urine or feces. Patterns of accumulation of these intermediates can distinguish between the different types of porphyria. Further confirmation by genetic testing is now widely available and facilitates screening of relatives. After diagnosis, management focusses on specific treatments, relief of symptoms, prevention of recurrences, surveillance for long-term complications and genetic counseling.
Introduction
This report provides a general overview of the types of porphyria. NORD has individual reports on each type.
ACUTE PORPHYRIAS
The acute (hepatic) porphyrias are characterized by attacks of abdominal pain (the most common symptom), constipation and other gastrointestinal symptoms, extremity pain and weakness and mental changes such as insomnia, anxiety, agitation, hallucinations, delusions and seizures. Examination during an attack often reveals rapid heart rate and hypertension. Attacks may last days or weeks. Other symptoms may include red or dark urine and burning or hesitancy with urination. Weakness can progress to generalized paralysis and the need for respiratory support. These severe symptoms can be life threatening especially if diagnosis and treatment are delayed. Effective treatments include intravenous infusion of heme (also known as hemin), mostly administered during acute attacks, and subcutaneous injection of givosiran, an interfering RNA therapeutic, which is effective for preventing frequently recurring attacks.
Acute Intermittent Porphyria (AIP)
Acute intermittent porphyria (AIP) is the most common of the acute porphyrias. Symptoms include periodic attacks as described above. Long-term complications of AIP and other acute porphyrias may include progressive kidney damage, high blood pressure and liver cancer. Symptoms begin during adulthood in almost all affected people and are more common in females. Photosensitivity occurs only in some individuals who develop advanced renal failure which can elevate levels of porphyrins in plasma.
People with AIP have a variant in the gene for the third enzyme in the pathway to make heme, which is porphobilinogen (PBGD) deaminase (PBGD), also known as hydroxymethylbilane synthase (HMBS). Inheritance is autosomal dominant (see Causes section). Most individuals who inherit a variant that can cause AIP never have symptoms and are said to have latent AIP. Others may have only one or a few attacks in their lifetimes. Unfortunately, a minority have attacks that recur frequently and chronic symptoms may persist between attacks. Many of the drugs, hormones and other factors associated with attacks of AIP and other acute porphyrias have the capacity to increase the synthesis of the first enzyme in the pathway to make heme in the liver, which is delta-aminolevulinic acid (ALA) synthase-1 (ALAS1) and this leads to accumulation of the porphyrin precursors ALA and PBG, especially during attacks. ALA and PBG may be neurotoxic, and their elevation are associated with neurological symptoms. The long-term risk of liver cancer is increased in AIP and other acute porphyrias compared to the general population.
Variegate Porphyria (VP)
Variegate porphyria (VP) is the second most common acute porphyria. Attacks are identical to those seen in AIP. Blistering skin lesions are more common than acute attacks and are often misdiagnosed as porphyria cutanea tarda (PCT), a much more common porphyria. People with VP have a variant in the gene for protoporphyrinogen oxidase (PPOX), the seventh enzyme in the heme synthetic pathway. As in AIP, the inheritance pattern is autosomal dominant, and symptoms develop after puberty in a minority of individuals with a disease-causing PPOX variant. Very rare individuals who inherit a PPOX variant from each parent have severe disease beginning early in life.
Hereditary Coproporphyria (HCP)
HCP is less common than VP but can cause the same symptoms. However, skin manifestations are much less common in HCP than in VP. People with HCP have a variant in the gene for coproporphyrinogen oxidase (CPOX), the sixth enzyme in the heme synthetic pathway. Inheritance is autosomal dominant and many individuals with a disease-causing CPOX variant never develop symptoms. Very rare individuals who inherit a CPOX variant from each parent have very severe symptoms beginning in childhood.
ALAD-Deficiency Porphyria (ADP)
ALAD porphyria (ADP) is extremely rare with only about 12 well documented cases reported. Inheritance is autosomal recessive, with a disease-causing ALAD variant inherited from each parent. Why almost all cases have been males is unexplained. Symptoms are mostly neurological, as in AIP, and may begin in childhood or during adolescence. Chronic neurological symptoms are seen in severe cases. In one case, onset in an adult was associated with a bone marrow disorder (polycythemia vera). ADP is classified as a hepatic porphyria but also has an erythropoietic component.
BLISTERING CUTANEOUS PORPHYRIAS
Porphyria Cutanea Tarda (PCT)
This hepatic porphyria is the most common of all porphyrias and is also the most readily treated. It is primarily an acquired, iron related disease in which there is inhibition of uroporphyrinogen decarboxylase (UROD), the fifth enzyme in the heme synthetic pathway in the liver. A minority or patients have a predisposing UROD gene variant inherited from one parent and are classified as having familial PCT. Symptoms usually occur after age 40. Sun-exposed areas of the skin (most commonly the backs of the hands) can become friable and prone to blistering, scarring and excess hair growth. Other predisposing factors include chronic hepatitis C, HIV infection, alcohol, smoking, estrogens and excess iron. Some of these factors and marked accumulation of porphyrins in the liver due to PCT itself can lead to chronic liver damage and liver cancer. In patients with chronic hepatitis C and PCT, the preferred treatment is an antiviral regimen, which are highly effective and can cure both conditions. Otherwise, PCT responds well to treatment with phlebotomies (to reduce iron) or a low dose regimen of hydroxychloroquine.
Erythropoietic Protoporphyria (EPP) and X-Linked Protoporphyria (XLP)
The photocutaneous symptoms of the protoporphyrias (EPP and XLP) usually begin in infancy or early childhood and are primarily acute and nonblistering. Sun exposure causes initial itching or mild burning, sometimes within minutes of exposure. More prolonged exposure leads to severe cutaneous pain, swelling and redness along with systemic symptoms that may last for several days. Affected people learn to avoid sunlight, so they seldom develop blistering or scarring but have restricted activities and impaired quality of life. Protoporphyrin is insoluble in water and is taken up by the liver and excreted in bile rather than urine. Excess protoporphyrin can cause significant liver damage but this occurs in less than 5% of patients. Current treatment is sunlight avoidance and agents that increase skin melanin pigmentation. Inheritance of EPP is autosomal recessive and due to inheritance of variants of the gene that encodes ferrochelatase (FECH) the last enzyme in the heme synthetic pathway. Most people with EPP have inherited a rare severe FECH variant from one parent and a common mild FECH variant (described as a low-expression or hypomorphic variant) from the other. The low expression variant is common (found in ~10% of Caucasians) but by itself causes no disease.
XLP results from a variant of the ALAS2 gene which encodes ALAS2, the form of the first enzyme in the pathway that is found exclusively in the bone marrow. The ALAS2 gene is located on the X chromosome so the inheritance pattern of XLP is X-linked. The ALAS2 variants in XLP increase ALAS2 enzyme activity and therefore are termed gain of function variants. The resulting overproduction of all intermediates in the heme synthetic pathway leads to the accumulation of protoporphyrin.
Congenital Erythropoietic Porphyria (CEP)
Congenital erythropoietic porphyria (CEP) is very rare, with onset usually in early childhood or even in utero. Inheritance is autosomal recessive and due to disease-causing variants of the gene that encodes uroporphyrinogen III synthase (UROS). UROS is the fourth enzyme in the heme synthetic pathway. Porphyrin levels in CEP are especially high and the blistering skin manifestations are usually severe. Repeated blistering, infection and scarring can lead to loss of fingers and facial features. Facial hair growth is often increased. Porphyrins are deposited in bones and teeth. Anemia in people with severe cases may require red blood cell transfusion. An enlarged spleen is common. Mild cases can resemble PCT but do not respond to treatments that are effective for that condition. Rare onset of CEP (and other erythropoietic porphyrias) during adulthood is often related to the development of a clonal bone marrow disease.
Hepatoerythropoietic Porphyria (HEP)
Hepatoerythropoietic porphyria (HEP) results from inheriting a UROD gene variant from each parent. HEP resembles CEP clinically, but unusually mild cases can resemble PCT. Symptoms usually begin in infancy or childhood.
As already noted, porphyrias occur when there is an alteration of one or more of the eight enzymes used to make the molecule heme. A different gene is responsible for making each of these enzymes. Each type of porphyria is related to variants in a different gene responsible for one of the enzymes involved in the steps making the molecule heme. Most variants cause decreased function of the affected enzyme, but variants in one type of porphyria (XLP) cause an increase in enzyme activity.
Porphyrias are inherited in families. However, many people who inherit a gene variant for one of the porphyrias have no symptoms. Especially in hepatic porphyrias, certain medications, chemical exposures or changes in diet may be necessary before someone with a gene variant for porphyria develops symptoms of the disease. Unknown modifying genes also have effects especially in hepatic porphyrias.
The following table summarizes the pattern of inheritance and the enzyme that is altered in each type of porphyria:
Autosomal Dominant Inheritance
Dominant genetic disorders occur when only a single copy of a disease-causing gene variant is necessary to cause the disease. The gene variant can be inherited from either parent or can be the result of a new (de novo) changed gene in the affected individual that is not inherited. The risk of passing the gene variant from an affected parent to a child is 50% for each pregnancy. The risk is the same for males and females.
Autosomal Recessive Inheritance
Recessive genetic disorders occur when an individual inherits a disease-causing gene variant from each parent. If an individual receives one normal gene and one disease-causing gene variant, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the gene variant and have an affected child is 25% with each pregnancy. The risk of having a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females. X-Linked Recessive Inheritance
X-Linked Inheritance
X-linked genetic disorders are conditions caused by a disease-causing gene variant on the X chromosome and mostly affect males. Females who have a disease-causing gene variant on one of their X chromosomes are carriers for that disorder. Carrier females usually do not have symptoms because females have two X chromosomes and only one carries the gene variant. Males have one X chromosome that is inherited from their mother and if a male inherits an X chromosome that contains a disease-causing gene variant, he will develop the disease.
Female carriers of an X-linked disorder have a 25% chance with each pregnancy to have a carrier daughter like themselves, a 25% chance to have a non-carrier daughter, a 25% chance to have a son affected with the disease and a 25% chance to have an unaffected son.
If a male with an X-linked disorder can reproduce, he will pass the gene variant to all his daughters who will be carriers. A male cannot pass an X-linked gene to his sons because males always pass their Y chromosome instead of their X chromosome to male children.
The exact number of people who have porphyria is unknown but it has been estimated that about 1 in 20,000 people may have some type of porphyria. Some forms of porphyria are more common in specific populations. For example, acute intermittent porphyria is more common in Sweden than other parts of the world. Porphyria cutanea tarda may be the most common type of porphyria, occurring in 1 in 25,000 people in the United States. Erythropoietic porphyria is the most common in children, with the highest incidence in the Netherlands of about 1 in 75,000.
Porphyria is diagnosed based on a clinical exam and symptoms, as well as blood, urine and stool tests. Molecular genetic (DNA) testing may be used to help confirm the diagnosis. Once someone in a family has been diagnosed with porphyria, other family members may be tested to see if they have inherited the condition.
Treatment
Effective treatment is available for most porphyrias. Porphyria cutanea tarda (PCT) is readily treated by phlebotomies (to reduce iron), low-dose hydroxychloroquine (to remove excess porphyrins from the liver) or by treating hepatitis C (if present). These treatments can normalize porphyrin levels and most people do not have recurrence of PCT.
Exacerbation of the acute porphyrias often requires hospitalization for management of severe symptoms. Hemin is approved by the U.S. Food and Drug Administration (FDA) to treat acute attacks. Glucose loading may be helpful but is less effective than hemin. Givosiran, a long-acting interfering RNA drug, is approved by the FDA and is effective for prevention of frequently recurring attacks. GnRH analogues that interrupt ovulation can be used to prevent attacks related to the menstrual cycle in women. Liver transplantation is an effective option for people with acute porphyria who become unresponsive to other treatments.
Afamelanotide (a human melanocyte stimulating hormone analogue) is approved by the FDA for treatment of protoporphyrias. It increases skin melanin and can greatly increase sun tolerance in people with EPP and XLP but does not lower circulating levels of protoporphyrin. People with protoporphyrias learn to avoid painful reactions by avoiding sunlight. However, people with blistering cutaneous porphyrias such as CEP have much less pain from sun exposure and are at risk for severe skin damage unless they learn to avoid sunlight. Marrow stem cell transplantation is effective in severe childhood cases of CEP.
Information on current clinical trials is posted on the Internet at https://clinicaltrials.gov/. All studies receiving U.S. Government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Tollfree: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website:
https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/.
For information about clinical trials sponsored by private sources, contact:
https://www.centerwatch.com/.
For information about clinical trials conducted in Europe, contact:
https://www.clinicaltrialsregister.eu/.
TEXTBOOKS
Phillips JD, Anderson KE. The porphyrias (Chapter 31). In: Lichtman MA, Prchal JT, Lichtman MA. eds. Williams Hematology: The Red Cell and Its Diseases, 1st edition. McGraw-Hill; 2022: pp 395-422.
Anderson KE. Acute Intermittent Porphyria, ALA-Dehydratase-Deficient Porphyria,
Congenital Erythropoietic Porphyria, Porphyria Cutanea Tarda, Variegate Porphyria and
Hereditary Coproporphyria. In: NORD Guide to Rare Disorders. Lippincott Williams &
Wilkins. Philadelphia, PA. 2003:490-95.
Mathews-Roth MM. Erythropoietic Porphyria. In: NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia, PA. 2003:495-96.
JOURNAL ARTICLES
Roach AN, Barkley H, Rodriquez C, Burrow TA, Anderson KE, Shukla A. Profound hypotonia in an infant with δ-aminolevulinic acid dehydratase deficient porphyria [published correction appears in Eur J Hum Genet. 2025 Mar 10. doi: 10.1038/s41431-025-01804-1.]. Eur J Hum Genet. Published online December 11, 2024. doi:10.1038/s41431-024-01758-w
Levy C, Dickey AK, Wang B, et al. Evidence-based consensus guidelines for the diagnosis and management of protoporphyria-related liver dysfunction in erythropoietic protoporphyria and X-linked protoporphyria. Hepatology. 2024;79(3):731-743. doi:10.1097/HEP.0000000000000546
Sardh E, Balwani M, Rees DC, Anderson KE, Jia G, Sweetser MT, Wang B. Long-term follow-up of givosiran treatment in patients with acute intermittent porphyria from a phase 1/2, 48-month open-label extension study. Orphanet Journal of Rare Diseases. 2024 Oct 3;19(1):365.
Bonkovsky HL, Rudnick SP, Ma CD, et al. Ledipasvir/Sofosbuvir Is Effective as Sole Treatment of Porphyria Cutanea Tarda with Chronic Hepatitis C. Dig Dis Sci. 2023;68(6):2738-2746. doi:10.1007/s10620-023-07859-8
Moghe A, Dickey A, Erwin A, Leaf RK, O’Brien A, Quigley JG, Thapar M, Anderson KE. Acute hepatic porphyrias: Recommendations for diagnosis and management with real-world examples. Molecular Genetics and Metabolism. 2023 Nov 1;140(3):107670. PMID: 37542766
Anderson KE, Lobo R, Salazar D, et al. Biochemical Diagnosis of Acute Hepatic Porphyria: Updated Expert Recommendations for Primary Care Physicians. Am J Med Sci. 2021;362(2):113-121. doi:10.1016/j.amjms.2021.03.004
Muschalek W, Hermasch MA, Poblete-Gutiérrez P, Frank J. The porphyrias. J Dtsch Dermatol Ges. 2022 Mar;20(3):316-331.
Marcacci M, Ricci A, Cuoghi C, Marchini S, Pietrangelo A, Ventura P. Challenges in diagnosis and management of acute hepatic porphyrias: from an uncommon pediatric onset to innovative treatments and perspectives. Orphanet J Rare Dis. 2022 Apr 7;17(1):160.
Gerischer LM, Scheibe F, Nümann A, Köhnlein M, Stölzel U, Meisel A. Acute porphyrias – a neurological perspective. Brain Behav. 2021 Nov;11(11): e2389. Epub 2021 Oct 17.
Di Pierro E, Granata F. Nutrients and porphyria: an intriguing crosstalk. Int J Mol Sci. 2020 May 14;21(10):3462.
Neeleman RA, Wensink D, Wagenmakers MAEM, Mijnhout GS, Friesema ECH, Langendonk JG. Diagnostic and therapeutic strategies for porphyrias. Neth J Med. 2020 Jul;78(4):149-160.
Yasuda M, Chen B, Desnick RJ. Recent advances on porphyria genetics: inheritance, penetrance & molecular heterogeneity, including new modifying/causative genes. Mol Genet Metab. 2019 Nov;128(3):320-331. Epub 2018 Nov 30.
Balwani M. Erythropoietic Protoporphyria and X-Linked Protoporphyria: pathophysiology, genetics, clinical manifestations, and management. Mol Genet Metab. 2019 Nov;128(3):298-303. Epub 2019 Jan 24.
Stölzel U, Doss MO, Schuppan D. Clinical guide and update on porphyrias. Gastroenterology. 2019 Aug;157(2):365-381.e4. Epub 2019 May 11.
Singal AK. Porphyria cutanea tarda: Recent update. Mol Genet Metab. 2019 Nov;128(3):271-281. Epub 2019 Jan 18.
Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. N Engl J Med. 2017 Aug 31;377(9):862-872.
INTERNET
Wang B, Bissell DM. Hereditary Coproporphyria. 2012 Dec 13 [Updated 2022 May 19]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK114807/ Accessed April 9, 2025.
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. Porphyria Cutanea Tarda, MIM Number: 176100: Last edited: 06/28/2022. World Wide Web URL: https://www.omim.org/entry/176100 Accessed April 9, 2025.
Erwin A, Balwani M, Desnick RJ; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Congenital Erythropoietic Porphyria. 2013 Sep 12 [Updated 2021 Apr 15]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK154652/ Accessed April 9, 2025.
Porphyria. National Digestive Diseases Information Clearinghouse. Last reviewed: July 2020. https://www.niddk.nih.gov/health-information/liver-disease/porphyria Accessed April 9, 2025.
Singal AK, Anderson KE. Variegate Porphyria. 2013 Feb 14 [Updated 2019 Dec 12]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK121283/ Accessed April 9, 2025.
Sardh E, Barbaro M. Acute Intermittent Porphyria. 2005 Sep 27 [Updated 2024 Feb 8]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1193/ Accessed April 9, 2025.
Porphyria. MedlinePlus. Medical Encyclopedia. Page last updated: March 20, 2017.
https://medlineplus.gov/porphyria.html Accessed April 9, 2025.
Rudnick S, Phillips J, Bonkovsky H; Porphyrias Consortium of the Rare Diseases Clinical Research Network. Familial Porphyria Cutanea Tarda. 2013 Jun 6 [Updated 2022 Jun 9]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK143129/ Accessed April 9, 2025.
Rudnick S, Phillips J, Bonkovsky H; Porphyrias Consortium of the Rare Diseases Clinical Research Network. Hepatoerythropoietic Porphyria. 2013 Oct 31 [Updated 2022 Dec 22]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK169003/ Accessed April 9, 2025.
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. Porphyria, Acute Porphyria, MIM number: 612740: Last edited: 02/27/2012. World Wide Web URL: https://www.omim.org/entry/612740 Accessed April 9, 2025.

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportPlease complete this form to access the requested resource.

