

Last updated:
9/25/2024
Years published: 1986, 1988, 1990, 1993, 1997, 1999, 2000, 2003, 2004, 2005, 2006, 2014, 2017, 2024
NORD gratefully acknowledges Gioconda Alyea, MD (FMG), MS, National Organization for Rare Disorders, Forbes D. Porter, MD, PhD, Senior Investigator, Clinical Director; Simona E. Bianconi, MD, Staff Clinician; and An N. Dang Do, MD, PhD, Staff Clinician; National Institute of Child Health and Human Development, Program in Developmental Endocrinology and Genetics, Section on Molecular Dysmorphology, for assistance in the preparation of this report.
Summary
Niemann-Pick disease type C (NPC) is a rare progressive genetic disorder characterized by an inability of the body to transport cholesterol and other fatty substances (lipids) inside of cells. This leads to the abnormal accumulation of these substances within various tissues of the body, including brain tissue. The accumulation of these substances damages the affected areas. NPC is highly variable, and the age of onset and specific symptoms can vary from one person to another, sometimes even among members of the same family. NPC can range from a fatal disorder within the first few months after birth (neonatal period) to a late onset, chronic progressive disorder that remains undiagnosed well into adulthood. Most cases are detected during childhood and progress to cause life-threatening complications by the second or third decade of life.
NPC is caused by changes (variants) in the NPC1 gene (NPC type 1C) or the NPC2 gene (NPC type 2C) and is inherited in an autosomal recessive manner.
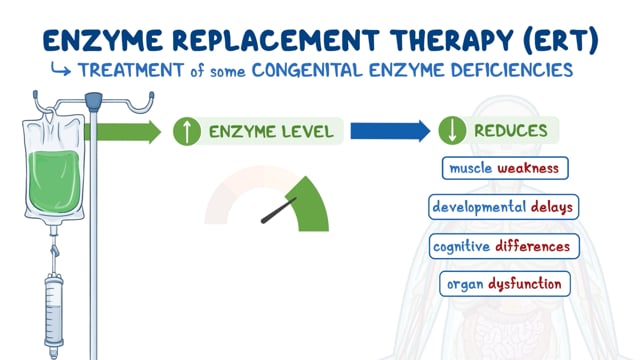
Recently, the U.S. Food and Drug Administration (FDA) approved arimoclomol (Miplyffa) in combination with the enzyme inhibitor miglustat to treat neurological symptoms associated with NPC in adults and children 2 years of age and older. Levacetylleucine (Aqneursa) has been FDA approved to treat the neurological symptoms caused by NPC in both adults and children.
Other treatments depend on the specific symptoms that the affected person has.
Introduction
NPC belongs to a larger group of more than 50 disorders known as lysosomal storage disorders. Lysosomes are membrane-bound compartments within cells. They contain enzymes that break down large molecules such as proteins, carbohydrates and fats into their building blocks. Abnormal functioning of a transport protein leads to the accumulation of cholesterol and other fatty substances in various tissues of the body, including brain tissue. NPC used to be grouped together with two other disorders, named Niemann-Pick disease type A and Niemann-Pick disease type B.
However, researchers have determined that the underlying defect in types A and B involves variants in the SMPD1 gene and deficiency of the enzyme acid sphingomyelinase, which does not occur in NPC. Niemann-Pick disease types A and B are now considered a distinct disorder called acid sphingomyelinase deficiency.
Individuals with NPC can have onset of symptoms at different ages that have been grouped historically as: perinatal (shortly before and after birth), early infantile (3 months to < 2 years), late infantile (2 to < 6 years), juvenile (6 to < 15 years) and adult (15 years and greater). NPC affects neurologic and psychiatric functions, as well as various internal organs (visceral). Symptoms arise at different times and follow independent progression. Visceral symptoms are more typically seen in individuals presenting at a younger age. Neurologic and psychiatric symptoms often occur slowly over time and thus feature more prominently in individuals presenting in the later age groups.
Because NPC is a highly variable disorder, it is important to note that affected individuals will not have all the symptoms described below and that every individual patient is unique. Some children will develop severe, life-threatening complications early in life; others have a mild disease that may go undiagnosed well into adulthood. Parents should talk to their child’s physician and medical team about the specific symptoms and overall prognosis.
The main symptoms according to age groups are the following:
Perinatal NPC (shortly before or after birth)
Early Infantile NPC (2 months to under 2 years)
Late Infantile NPC (2 to under 6 years)
Juvenile NPC (6 to 15 years)
Adolescent and Adult NPC (15 years and older)
Neurological and psychiatric symptoms (common across ages)
End-stage disease
Some older adults may first be misdiagnosed with dementia or psychiatric illness such as major depression or schizophrenia. Individuals have been described in medical literature with other psychiatric manifestations such as obsessive-compulsive disorder, bipolar disorders and hallucinations.
Following a long term gradual neurological decline death often results from aspiration pneumonia and subsequent respiratory failure, or intractable epilepsy not responding to medical intervention.
Individuals with NPC have variants in the NPC1 or NPC2 genes. Approximately 95% of affected individuals have variants in NPC1. Genes provide instructions for producing proteins that play a critical role in many functions of the body. Variants in a gene may lead to the production of a protein that has reduced or abnormal functions, or to the absence of the protein. Depending upon the functions of the protein, this can affect many organ systems of the body, including the brain.
The exact function of the NPC1 and NPC2 proteins is not fully understood. Researchers do know that the protein products of these genes are involved in the movements (trafficking) of large molecules within cells. When variants in the NPC1 or NPC2 gene occur, insufficient levels of functional protein products are made. This causes abnormal accumulation of cholesterol in the peripheral tissues of the body such as the liver and spleen, and accumulation of cholesterol and glycosphingolipids (complex compounds consisting of fatty material and carbohydrates) in the brain. The accumulation of these materials causes the various observable symptoms of NPC.
NPC follows autosomal recessive inheritance. Recessive genetic disorders occur when an individual inherits a disease-causing gene variant from each parent. If an individual receives one normal gene and one disease-causing gene variant, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the gene variant and have an affected child is 25% with each pregnancy. The risk of having a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
NPC affects males and females in equal numbers and can affect individuals of any ethnic background (pan ethnic). NPC is estimated to occur in 1 in 100,000-120,000 live births. However, many cases go misdiagnosed or undiagnosed, making it difficult to determine the disorder’s true frequency in the general population.
Niemann-Pick disease type C is diagnosed based on characteristic symptoms obtained from a thorough clinical evaluation (see under Signs and Symptoms) and confirmed by a variety of specialized tests. Proper diagnosis of NPC requires physicians to suspect the diagnosis based upon symptoms, and to follow up with appropriate laboratory tests to evaluate the function of the protein or the presence of accumulated byproducts (biochemical tests and to identify variants in the NPC1 or NPC2 genes.
Many physicians have little experience with NPC. Thus, affected individuals and families often face a significant delay in diagnosis. Clinical experts on NPC have developed a Suspicion Index Tool based on visceral, neurologic and psychiatric symptoms to help physicians unfamiliar with the disorder to diagnose NPC. Further study and refinement of the Suspicion Index Tool is necessary to determine its usefulness in clinical practice.
Specific signs and symptoms that may allow to diagnose the condition early include:
In children, signs like an enlarged liver or spleen (hepatosplenomegaly), problems with bile flow (cholestasis), or fluid buildup in the abdomen (ascites), combined with neurological signs like ataxia or VSSP, could be an indication of NPC.
Clinical Testing and Workup
Traditionally the confirmation of an NPC diagnosis was done by staining the affected individual’s skin cells (fibroblasts) for level of cholesterol accumulation (filipin staining). Testing for the cells’ ability to modify cholesterol (cholesterol esterification test) has also been used for diagnostic purposes.
Recently, blood-based testing for biomarkers (oxysterols, lysosphingolipids, bile acid metabolites) and genetic testing with molecular gene sequencing of the NPC1 and NPC2 genes have replaced these traditional methods.
Filipin test is no longer recommended as a first line test for the diagnosis of NPC, but it is a useful diagnostic tool in uncertain cases wherein the biomarkers and/or molecular analysis is inconclusive. Genetic testing is readily available for NPC, for both NPC1 and NPC2 genes. In clinical practice, it is generally used as a confirmatory test after initial screening by measuring levels of oxysterols. The level of oxysterols is only done when clinical features suggest the diagnosis of NPC.
Once the diagnosis is confirmed, special attention should be paid to look for disease spread and manifestations of the disease.
The most common neurological manifestation in NPC is cerebellar ataxia, vertical supranuclear gaze palsy (VSGP), dysarthria, dysphagia, dementia and movement disorders.
Treatment
Treatment of NPC may require the coordinated efforts of a team of specialists. Pediatricians, neurologists, ophthalmologists, pulmonologists, gastroenterologists, and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Psychosocial support for the entire family is essential as well. Genetic counseling would benefit affected individuals and their families.
Studies have shown that the medication miglustat (Zavesca) may also be able to slow the progression of neurological symptoms associated with NPC. Miglustat blocks the synthesis of glycosphingolipids, one of the substances that accumulates in the brain of individuals with NPC. Together, Miplyffa and miglustat offer a more comprehensive approach to managing the neurological effects of NPC.
In 2024, arimoclomol (Miplyffa) in combination with the enzyme inhibitor miglustat was approved by the U.S. Food and Drug Administration (FDA) to treat neurological symptoms associated with NPC in adults and children 2 years of age and older.
In 2024, the FDA approved a new drug called levacetylleucine (Aqneursa) for treating the neurological symptoms caused by NPC in both adults and children who weigh at least 15 kilograms (about 33 pounds).
Other treatments depend on the specific symptoms that the affected people have:
Swallowing difficulties (dysphagia) are common in NPC. These should be regularly monitored to prevent complications like aspiration. Initial treatment may involve softening solids and thickening liquids, and a speech therapist can help improve swallowing function. In advanced cases, a gastronomy tube may be needed to provide nutrition directly into the stomach.
Seizures are another major symptom and may respond to anti-seizure medications (antiepileptics). However, in later stages of the disease, seizures may become resistant to treatment.
Cataplexy (sudden muscle weakness) can be managed with certain medications, such as tricyclic antidepressants or central nervous system stimulants. For issues like dystonia and tremors, anticholinergic agents or botulinum toxin injections may be helpful.
Sleep disorders are common, with some individuals experiencing insomnia, fragmented sleep, or conditions like obstructive sleep apnea. Treatments for sleep issues might include melatonin, nocturnal sedatives, or in severe cases of sleep apnea, a CPAP machine may be necessary to keep the airways open during sleep.
Many people affected with NPC benefit from an individualized educational plan that includes physical therapy, speech therapy and occupational therapy. Resources for respite care and support for family members are also important in managing the challenges of the disease.
NPC specifically affects cholesterol processing within the cells, so diets low in fats and cholesterol do not impact the progression of the neurological symptoms.
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Toll-free: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website:
https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, in the main, contact:
www.centerwatch.com
For more information about clinical trials conducted in Europe, contact: https://www.clinicaltrialsregister.eu/
RareConnect offers a safe patient-hosted online community for patients and caregivers affected by this rare disease. For more information, visit www.rareconnect.org.
TEXTBOOKS
Patterson MC. Niemann-Pick Disease, Type C. In: NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia, PA. 2003:485.
JOURNAL ARTICLES
Labrecque M, Touma L, Bhérer C, et al. Estimated prevalence of Niemann–Pick type C disease in Quebec. Sci Rep. 2021;11: 22621. https://www.nature.com/articles/s41598-021-01966-0
Geberhiwot T, Moro A, Dardis A, et al. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J Rare Dis. 2018; 13: 50. https://ojrd.biomedcentral.com/articles/10.1186/s13023-018-0785-7
Mengel E, Pineda M, Hendriksz CJ, Walterfang M, Torres JV, Kolb SA. Differences in Niemann-Pick disease Type C symptomatology observed in patients of different ages. Mol Genet Metab. 2017;120(3):180-189. doi:10.1016/j.ymgme.2016.12.003.
Papandreou A, Gissen P. Diagnostic workup and management of patients with suspected Niemann-Pick type C disease. Therapeutic Advances in Neurological Disorders 2016;9(3):216-229.
Pineda M, Mengel E, Jahnova H, Heron B, Imrie J, Lourenco CM, van der Linden V, Karimzadeh P, Valayannopoulos V, Jesina P et al. A suspicion index to aid screening of early-onset Niemann-Pick disease type C (NP-C). BMC Pediatrics 2016;16:107.
Vanier MT, Gissen P, Bauer P, Coll MJ, Burlina A, Hendriksz CJ, Latour P, Goizet C, Welford RW, Marquardt T et al. Diagnostic tests for Niemann-Pick disease type C (NP-C): A critical review. Molecular Genetics and Metabolism 2016;118(4):244-254.
Walkley SU, Davidson CD, Jacoby J, Marella PD, Ottinger EA, Austin CP, Porter FD, Vite CH, Ory DS. Fostering collaborative research for rare genetic disease: the example of Niemann-Pick type C disease. Orphanet Journal of Rare Diseases 2016;11(1):161.
Alobaidy H. Recent advances in the diagnosis and treatment of Niemann-Pick disease type C in children: a guide to early diagnosis for the general pediatrician. International Journal of Pediatrics. February 16, 2015. https://onlinelibrary.wiley.com/doi/10.1155/2015/816593
Santos-Lozano A, Villamandos Garcia D, Sanchis-Gomar F, Fiuza-Luces C, Pareja-Galeano H, Garatachea N, Nogales Gadea G, Lucia A. Niemann-Pick disease treatment: a systematic review of clinical trials. Annals of Translational Medicine 2015; 3(22):360.
Wraith JE, Sedel F, Pineda M, Wijburg FA, Hendriksz CJ, Fahey M, Walterfang M, Patterson MC, Chadha-Boreham H, Kolb SA. Niemann-Pick type C suspicion index tool: analyses by age and association of manifestations. Journal of Inherited Metabolic Disease 2014;37(1):93-101.
Patterson MC, Mengel E, Wijburg FA, Muller A, Schwierin B, Drevon H, Vanier MT, Pineda M: Disease and patient characteristics in NP-C patients: findings from an international disease registry. Orphanet Journal of Rare Diseases 2013; 8:12.
Jiang X, Sidhu R, Porter FD, Yanjanin NM, Speak AO, te Vruchte DT, Platt FM, Fujiwara H, Scherrer DE, Zhang J et al. A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. Journal of Lipid Research 2011;52(7):1435-1445.
INTERNET
Patterson M. Niemann-Pick Disease Type C. 2000 Jan 26 [Updated 2020 Dec 10]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1296/ Accessed Sept 25, 2024.
Schiffmann R. Overview of Niemann-Pick disease. UpToDate. March 12, 2024. https://www.uptodate.com/contents/overview-of-niemann-pick-disease Accessed Sept 25, 2024.

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportPlease complete this form to access the requested resource.

