

Last updated:
July 16, 2020
Years published: 1986, 1987, 1988, 1990, 1995, 1996, 1999, 2000, 2005, 2014, 2017, 2020
NORD gratefully acknowledges Professor Roberto Giugliani, MD, PhD, Medical Genetics Service, HCPA, and Department of Genetics, UFRGS, Porto Alegre, Brazil, for assistance in the preparation of this report.
Summary
Maroteaux-Lamy syndrome (mucopolysaccharidosis type VI; MPS VI) is a rare genetic disorder characterized by complete or partial lack of activity of the enzyme arylsulfatase B (also called N-acetylgalactosamine-4-sulfatase), encoded by the ARSB gene. Deficiency or absence of this enzyme activity leads to the accumulation of complex carbohydrates called glycosaminoglycans (previously known as mucopolysaccharides) in the body. Abnormal accumulation of mucopolysaccharides leads to progressive involvement of multiple organ systems. The symptoms and severity of Maroteaux-Lamy syndrome can vary dramatically from one person to another; some individuals only develop mild symptoms, while others develop severe, even life-threatening complications. Common symptoms can include coarse facial features, corneal clouding, joint abnormalities, various skeletal malformations, an abnormally enlarged liver and/or spleen (hepatosplenomegaly), and hearing loss. Cardiac disease and restrictive pulmonary disease can also occur. Intelligence is usually not affected. In 2005, the Food and Drug Administration (FDA) approved the enzyme replacement therapy known as Naglazyme® for the treatment of Maroteaux-Lamy syndrome. Maroteaux-Lamy syndrome occurs due to mutations in the ARSB gene and is inherited as an autosomal recessive disorder.
Introduction
The mucopolysaccharidoses (MPS) are a group of inherited lysosomal storage disorders. More than 60 lysosomal storage disorders have been identified so far. Lysosomes function as the primary digestive units within cells. Enzymes within lysosomes break down or digest particular metabolites, such as certain carbohydrates and fats. In individuals with MPS disorders, deficiency or malfunction of specific lysosomal enzymes leads to an abnormal accumulation of certain complex carbohydrates known as mucopolysaccharides or glycosaminoglycans in the arteries, skeleton, eyes, joints, ears, skin, and/or teeth. These accumulations may also be found in the respiratory system, liver, spleen, central nervous system, blood, and bone marrow, causing progressive damage to cells, tissues, and various organ systems of the body. There are several different types and subtypes of MPS. These disorders, with one exception (MPS type II), are inherited in an autosomal recessive manner. Maroteaux-Lamy syndrome (mucopolysaccharidosis type VI, or MPS VI) was named from the two French physicians who first described this disorder in the medical literature in 1963.
The symptoms, onset and rate of progression of Maroteaux-Lamy syndrome vary greatly from one person to another. The disorder can be thought of as a spectrum or continuum of disease. Some individuals may only have a few symptoms and others may have serious symptoms affecting several different organ systems simultaneously. Maroteaux-Lamy syndrome can potentially cause life-threatening complications. Some individuals will have severe symptoms during infancy, while others have slowly progressive symptoms that develop over the course of multiple decades.
The variable nature of Maroteaux-Lamy syndrome means that most affected individuals will not have all of the symptoms potentially associated with the disorder. Individuals with this disorder can differ from one another dramatically. Parents should talk to their children’s physician and medical team about their child’s specific case, associated symptoms and overall prognosis. Most affected individuals come to medical attention during middle childhood.
Rapidly progressive Maroteaux-Lamy syndrome is associated with an onset of symptoms before three years of age. Affected individuals may develop walking problems (impaired mobility) by the age of 10 and experience delayed or absence of puberty. These individuals may be at risk of heart failure by second or third decades of life.
Slowly progressive disease is characterized by later onset. A diagnosis is usually obtained after five years of age, most often during the second or third decade. Despite a slower progression, individuals may still develop a decrease in overall function and ability by their late teen-aged years. Most individuals with Maroteaux-Lamy syndrome will develop serious complications at some point such as joint degeneration, cardiovascular disease, reduced pulmonary function or sleep apnea.
Intelligence is usually not affected in Maroteaux-Lamy syndrome. However, learning difficulties may be present as a consequence of other problems associated with the disorder (e.g., hearing loss).
Affected children may also exhibit failure to thrive and difficulty feeding. Short stature occurs in almost all patients and is described as disproportionate because the trunk may be shorter than the legs. In severe cases, final height may be less than 4 feet (120 centimeters). Degenerative joint disease is also common and can lead to the development of multiple joint contractures. A contracture occurs when thickening or shortening of tissue such as muscle fibers cause deformity and restrict the movement of an affected joint.
Individuals with Maroteaux-Lamy syndrome may be described as having ‘dysostosis multiplex’ a group of skeletal abnormalities that can be seen on x-ray examination. These abnormalities include thickened, short bones of the palm of the hands (metacarpals), underdeveloped (hypoplastic) and irregular wrist bones (carpal bones), abnormal ankle bones (tarsal bones), malformation (dysplasia) of the head of the thighbone (dysplastic femoral head), severe malformation of the hip, abnormalities of the ribs and spine, thickened collarbones (clavicles), and underdevelopment of the bones of the forearm (ulna and radius). Additional skeletal malformations may include a prominent breastbone (pectus carinatum), abnormal curvature of the spine, and knock-knees (genu valgum).
Skeletal malformations can be associated with a variety of complications. Affected individuals may develop pain, especially of the joints and hip, spinal cord compression, an abnormal manner of walking (gait), or difficulty walking. Affected joints may have a limited range of motion making daily tasks difficult. For example, the ability to fully move the shoulders may make simple tasks such as putting on a shirt or combing hair difficult.
Distinctive facial features usually do not occur in individuals with mild forms of the disorder. Individuals with severe forms often share distinctive facial characteristics and these individuals may resemble one another in facial appearance. Such characteristics include chubby faces, thickened lips due to the overgrowth of the gums (gingival hypertrophy), an unusually prominent forehead (frontal bossing), and a broad, flattened bridge of the nose. In some affected individuals, the tongue may be enlarged (macroglossia). Abnormal growth of hair on the face or the body may also occur (hirsutism). Some individuals have a short, stiff neck.
Clouding (opacity) of the thin transparent covering of the front of the eye (cornea) may also occur. If corneal clouding is severe the patient may present vision loss, particularly in dim light. Some individuals may sensitive to bright lights. If the nerve-rich lining the back of the eyes (retina) is involved, affected individuals may have reduced peripheral vision or develop night blindness. In some cases, increased pressure within the eye (glaucoma) may also develop. Increased intraocular pressure may cause “thinning, cupping, or notching of the disc rim. Less commonly, additional eye abnormalities may occur including degeneration of the nerve that transmits visual information from the retina to the brain (optic nerve atrophy).
Affected individuals may experience chronic watery, mucous discharge from nose (rhinorrhea), frequent sinus infections, and middle ear infections (otitis media). Hearing loss is common. Hearing loss may be due to failure of sound to be conducted from the outer ear trough the eardrum and tiny bones of the middle ear (conductive hearing loss) or may be due to damage to the inner ear or the nerves that transmit sound from the inner ear to the brain (sensorineural). In some cases, hearing loss may be due to a combination of both problems (mixed hearing loss).
Abnormalities of the heart are common in children with Maroteaux-Lamy syndrome. Symptoms associated with heart disease can include breathlessness, cough, wheezing, excessive sweating and recurrent chest infections. High blood pressure (hypertension) may also occur. Cardiac abnormalities can include narrowing (stenosis) and insufficiency of certain valves of the heart including the aortic, the mitral, and the tricuspid valves. Heart valves ensure that blood flows in only one direction within the heart. When a valve is damaged or malformed, blood can flow backward from one chamber back into another. Slowly progressive valvar heart disease can be present for years without causing symptoms. Eventually, valvar heart disease can cause a heart murmur. Narrowing of the heart valves can progressively make it more difficult for the heart to pump blood and can eventually result in heart failure.
Additional heart abnormalities can include disease or weakening of the heart muscle (cardiomyopathy) and endocardial fibroelastosis. Cardiomyopathy can be associated with a progressive inability to pump blood, fatigue, heart block, and fast heartbeats (arrhythmia). Endocardial fibroelastosis is a condition characterized by thickening of the endocardium of the ventricles. The endocardium is the innermost layer of tissue that surrounds the heart. These conditions can make it more difficult for the heart to pump blood effectively and can eventually cause heart failure and sudden cardiac death.
The lungs and other parts of the pulmonary system are usually affected. The storage of mucopolysaccharides may cause affected tissue to swell, which can obstruct various airways in the body, resulting in a high-pitched, loud respiratory sound (stridor) and airway compromise. Thick, mucous secretions can further clog the airways. The chest may become rigid, preventing the lungs from taking in sufficient amounts of air. Obstructive and restrictive lung disease can cause breathlessness, reduced endurance, recurrent episodes of pneumonia, and/or sleep apnea. Some affected infants have abnormal softening and weakening of the cartilage of the trachea (windpipe) so that the walls of the trachea are floppy instead of rigid (tracheomalacia). This is often mild, but can be severe, leading to collapse of the air passage. Tracheomalacia can contribute to breathing difficulties and may precipitate respiratory arrest. In some cases, the tonsils and adenoids may become enlarged narrowing the airway in the throat and contributing to breathing difficulties.
Abnormal enlargement of the liver (hepatomegaly) is common in individuals with Maroteaux-Lamy syndrome. The spleen may also be enlarged (splenomegaly). Hernias, conditions in which the abdominal membrane or contents protrude through a weak point in the abdominal wall, are also common. Umbilical hernias occur when the contents protrude from behind the bellybutton; inguinal hernias occur in the groin area. Some affected individuals have a protruding or bulging abdomen because of weakened muscles and/or hepatosplenomegaly.
Some affected individuals may develop hydrocephalus, a condition in which the accumulation of excess cerebrospinal fluid in the skull causes increased pressure on the brain, potentially causing a variety of signs and symptoms, including headache and/or papilledema.
Another common symptom associated with Maroteaux-Lamy syndrome is carpal tunnel syndrome, a condition caused by compression of a nerve running through the wrist. Symptoms usually begin as chronic tingling, burning or numbness in the wrist. Eventually, it can progress to cause sharp, piercing pain. Less often, tarsal tunnel syndrome, a similar condition affecting the ankle, may also occur.
Certain gastrointestinal symptoms including loose stools, diarrhea, or severe constipation have also been reported in individuals with Maroteaux-Lamy syndrome.
Maroteaux-Lamy syndrome is caused by changes (mutations) in the ARSB gene. Genes provide instructions for making proteins that play a critical role in many functions of the body. When a mutation of a gene occurs, the protein product may be faulty, inefficient, or absent. Depending upon the functions of the particular protein, this can affect many organ systems of the body.
The ARSB gene is related to (encodes) the lysosomal enzyme arylsulfatase B. Deficiency of this enzyme results in the accumulation of dermatan sulfate and chondroitin sulfate in the cells of various tissues because the body cannot breakdown glycosaminoglycans. More than 220 different mutations in the ARSB gene have been identified. Certain mutations are more likely to be associated with specific symptoms and/or severity (genotype-phenotype correlation).
The ARSB mutation is inherited as an autosomal recessive disorder. Genetic diseases are determined by the combination of alleles for a particular gene that are on the chromosomes received from the father and the mother. Autosomal recessive genetic disorders occur when it is needed that the individual presents a pathogenic mutation in each copy (allele) of the gene, being one allele inherited from the father, and the other allele inherited from the mother. To have the disease the individual should inherit two mutated copies of the same gene, one from each parent. If an individual receives one normal copy of the gene and another copy with a mutation that causes the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% in each pregnancy. The risk to have a child who is a carrier like the parents is 50% in each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Maroteaux-Lamy syndrome affects males and females in equal proportion. The prevalence of all forms of MPS is estimated to be 1 in 65,000 births. Although the exact incidence and prevalence of the disorder is unknown, it is estimated to occur in one case in 250.000 to 600,000 individuals. In some areas, due to founder effect and endogamy, prevalence is higher. However, we should take into account that MPS disorders, especially milder forms, often go unrecognized, so they may be underdiagnosed or misdiagnosed, making it difficult to determine their true frequency in the general population.
A diagnosis of Maroteaux-Lamy syndrome is based upon identification of characteristic symptoms, a detailed patient and family history, a thorough clinical evaluation and a variety of specialized tests.
Clinical Testing and Workup
In individuals suspected of Maroteaux-Lamy syndrome the urine could be initially examined to assess the levels of the glycosaminoglycans (GAGs). Elevated levels of total urinary GAGs, with predominance of dermatan sulfate, suggests Maroteaux-Lamy syndrome. To confirm the diagnosis, a blood sample should be taken to measure the activity of the enzyme arylsulfatase B. Deficient or absent activity of this enzyme is confirms the diagnosis of Maroteaux-Lamy syndrome. Although less convenient, the enzyme activity can be also measured in certain types of skin cells (fibroblasts). When a deficiency of arylsulfatase B enzyme is detected, at least another sulfatase should be measured to rule out multiple sulfatase deficiency. In Maroteaux-Lamy syndrome the only enzyme deficient will be arylsulfatase B, while in multiple sulfatase deficiency several sulfatases have low activity.
Whenever possible the sequencing of ARSB gene should be performed to identify the causative mutations. Although not mandatory to confirm the diagnosis, the identification of the mutations could help in the prediction of the clinical severity, in the identification of carriers in the family and prenatal diagnosis in future at-risk pregnancies.
The quantification of dermatan sulfate levels, the measurement of the activities of arylsulfatase A and other sulfatases, and gene sequencing, can also be performed in dried blood spots, a very convenient sample that is becoming increasingly used. This makes technically feasible to include the detection of this disease in newborn screening panels.
Treatment
The treatment of Maroteaux-Lamy syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, surgeons, orthopedists, cardiologists, dental specialists, ear-nose-throat specialists (otorhinolaryngologists), specialists who deal with diseases of the lungs and respiratory tract (pulmonologists), specialists who asses and treat hearing problems (audiologists), specialists who asses and treat vision problems (ophthalmologists), and other healthcare professionals may need to systematically and comprehensively plan the treatment. Genetic counseling may be of benefit for affected individuals and their families. Psychosocial support for the entire family is essential as well.
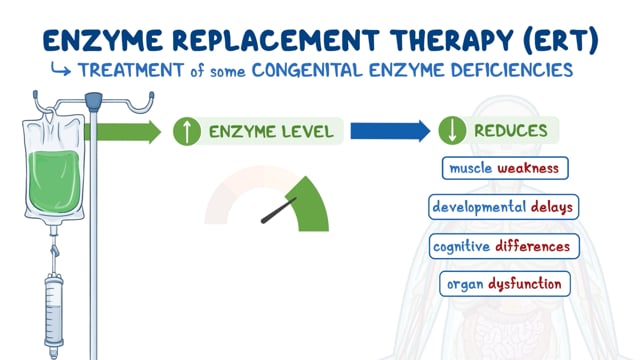
In 2005, the U.S. Food and Drug Administration (FDA) approved the orphan drug Naglazyme (galsulfase) for the treatment of individuals with Maroteaux-Lamy syndrome. Naglazyme is an enzyme replacement therapy (ERT), a therapy in which the missing or inactive enzyme is replaced with a genetically engineered (recombinant) version. Studies on long-term follow-up of ERT with galsulfase are now available, and indicate an acceptable safety profile with several improvements demonstrated, including extended survival.
Additional treatment is symptomatic and supportive. For example, surgery may be necessary to treat various abnormalities associated with Maroteaux-Lamy syndrome including carpal tunnel syndrome, skeletal malformations, spinal cord compression, degeneration of the hip, and hernias. Heart valve replacement may be necessary in some cases. Surgical removal of the tonsils and/or adenoids may be recommended. Tracheomalacia is usually treated by noninvasive measures, but in rare cases can require surgical intervention.
Hydrocephalus may be treated by the insertion of a tube (shunt) to drain excess of cerebrospinal fluid (CSF) away from the brain and into another part of the body where the CSF can be absorbed. A corneal transplantation can be performed for individuals with severe corneal clouding. Individuals with conductive hearing loss may experience the accumulation of a sticky fluid within the middle ear (glue ear). This is treated with a procedure called a myringotomy, in which a thin incision is made in the eardrum to release the fluid. There is no specific treatment for sensorineural hearing loss. Hearing aids may help to maximize remaining hearing.
Certain medications can be used to treat heart abnormalities, asthma-like episodes, and chronic infections. Anti-inflammatory medications may be of benefit. Respiratory insufficiency may require treatment with supplemental oxygen. Aggressive management of airway secretions is necessary as well.
Some affected individuals may undergo a sleep study, in which people are evaluated on how well they sleep and how well their body responds to sleep problems. Sleep apnea may be treated with continuous positive airway pressure (CPAP), which involves the use of a mask or similar device to deliver mild air pressure to keep airways open. In some cases, a similar treatment method known as bilevel positive airway pressure (BPAP) may be used. BPAP devices provide more pressure when you breathe in and less pressure when you breathe out.
Physical therapy and exercise may improve joint stiffness. Speech therapy may help children with hearing loss communicate effectively. Nutritional counseling and occupational therapy may also beneficial.
Hematopoietic stem cell transplantation (HSCT) as a way to replace the defective enzymes has been studied as a treatment for individuals with mucopolysaccharidoses. Hematopoietic stem cells are specialized cells found in the bone marrow (the soft spongy material found in the long bones). These blood stem cells grow and eventually develop into one of the three main types of blood cells — red cells, white cells, or platelets. A transplant is done to replace the bone marrow (and consequently the whole blood system) of an affected individuals with marrow from a person who does not have a particular disorder. The healthy cells produced by the new marrow contain sufficient levels of the lysosomal enzyme arylsulfatase B. The effectiveness of HSCT has varied greatly. Physical characteristics may improve, such as cloudy corneas may become clear, the size of an enlarged liver or spleen may be reduced, and mucopolysaccharides levels may drop. Some other symptoms are unaffected. The effect on neurological symptoms, such as cervical cord compression, varies considerably. HSCT in Maroteaux-Lamy syndrome has been carried out in a small number of patients. Because HSCT is a procedure that carries significant risks, it should be considered in selected cases, after careful evaluation. More research is necessary to determine the long-term safety and effectiveness of HSCT for individuals with Maroteaux-Lamy syndrome.
Pentosan poly-sulfate (PPS), a drug that has anti-inflammatory and pro-chondrogenic properties, was tested in MPS I, leading to reduction in urinary GAGs and improvement in joint mobility and pain scores. It can potentially also benefit patients with Maroteaux-Lamy syndrome, as indicated in preclinical studies.
Trials with a small molecule (Odiparcil) that promote clearance of intracellular GAGs are in progress in patients with Maroteaux-Lamy syndrome, and can result in add-on drug therapies to improve some areas where the benefits of enzyme replacement therapy are limited (cornea, heart valves, and others).
The use of losartan to improve some cardiovascular manifestations, particularly aortic root dilatation, is also being tested in a clinical trial in patients with Maroteaux-Lamy syndrome.
Gene therapy is also being studied as another approach to therapy for individuals with mucopolysaccharidoses such as Maroteaux-Lamy syndrome. In gene therapy, the defective gene present in a patient is replaced with a normal gene to enable the production of the active enzyme and prevent the development and progression of the disease. Given the permanent transfer of the normal gene, which is able to produce active enzyme at all sites of disease, this form of therapy is theoretically most likely to lead to a ‘cure’. A trial with the intravenous administration of an AAV vector that carry a normal sequence of the ARSB gene is in progress.
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Toll-free: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website:
https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, in the main, contact:
https://www.centerwatch.com
For more information about clinical trials conducted in Europe, contact: https://www.clinicaltrialsregister.eu/
TEXTBOOKS
Giugliani R. The mucopolysaccharidoses. In: Lysosomal storage siseases: a practical guide (eds: Mehta A, Winchester B). Wiley-Blackwell, Hoboken, NJ, USA 2013: 94-100.
Clarke JTR. Mucopolysaccharide Storage (MPS) Diseases. In: NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia, PA. 2003:474-476.
Gorlin RJ, Cohen MMJr, Hennekam RCM. Eds. Syndromes of the Head and Neck. 4th ed. Oxford University Press, New York, NY; 2001:135-137.
JOURNAL ARTICLES
Akyol MU, Alden TD, Amartino H, et al – Recommendations for the management of MPS VI: systematic evidence- and consensus-based guidance. Orphanet J Rare Dis 2019;14(1): 118. doi: 10.1186/s13023-019-1080-y.
Tomanin R, Karageorgos L, Zanetti A, et al. Mucopolysaccharidosis type VI (MPS VI) and molecular analysis: review and classification of published variants in the ARSB gene. Hum Mutat 2018; 39(12):1788-1802. doi: 10.1002/humu23613.
Quartel A, Harmatz PR, Lampe C, et al. Long-term galsulfase treatment associated with improved survival of patients with mycopolysaccharidosis VI (Maroteaux-Lamy syndrome): 15-year follow-up from the survey study. J Inborn Errors Metab Screen 2018; 6:1-6. doi: 10:1177/2326409818755800.
Harmatz PR, Shediac R. Mucopolysaccharidosis VI: Pathophysiology, diagnosis and treatment. Front Biosci 2017; 22: 385-406. Doi: 10.2741/4490.
Harmatz PR, Hendriksz CJ, Lampe C, et al. The effect of galsulfase enzyme replacement therapy on the growth of patients with mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). Mol Genet Metab 2017; 122(102): 107-112. doi: 10.1016/j.ymgme.2017.03.008.
Giugliani, R, Lampe C, Guffon N, et al. Natural history of galsulfase treament in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome) – 10 year follow-up of patients who previously participated in an MPS VI survey study. Am J Med Genet 2019; 164A (8):1953-64. doi: 10.1002/ajmg.a.36584.
Kampmann C, Lampe C, Whybra-trümpler C, et al. Mucopolysaccharidosis VI: cardiac involvement and the impact of enzyme replacement therapy. J Inherit Metab Dis. 2014;37(2):269-76. https://www.ncbi.nlm.nih.gov/pubmed/24062198
Jester S, Larsson J, Eklund EA, et al. Haploidentical stem cell transplantation in two children with mucopolysaccharidosis VI: clinical and biochemical outcome. Orphanet J Rare Dis. 2013;8:34. https://www.ncbi.nlm.nih.gov/pubmed/24107440
Brands MM, Oussoren E, Ruijter GJ. Up to five years experience with 11 mucopolysaccharidosis type VI patients. Mol Genet Metab. 2013;109:70-76. https://www.ncbi.nlm.nih.gov/pubmed/23523338
Brands MM, Hoogeveen-Westerveld M, Kroos MA, et al. Mucopolysaccharidosis type VI phenotypes-genotypes and antibody response to galsulfase. Orphanet J Rare Dis. 2013;8:51. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3637222/
Braunlin E, Rosenfeld H, Kampmann C, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: long-term cardiac effects of galsulfase (Naglazyme®) therapy. J Inherit Metab Dis. 2013;36:385-394. https://www.ncbi.nlm.nih.gov/pubmed/22669363
Jurecka A, Golda A, Opoka-Winiarska V, et al. Mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome) with a predominantly cardiac phenotype. Mol Genet Metab. 2011;104:695-699. https://www.ncbi.nlm.nih.gov/pubmed/21917494
Valayannopoulos V, Nicely H, Harmatz P, Tuberville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5. https://www.ojrd.com/content/5/1/5
Decker C, Yu ZJ, Giugliani R, et al. Enzyme replacement therapy for mucopolysaccharidosis VI: growth and pubertal development in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase. J Pediatr Rehabil Med. 2010;3:89-100. https://www.ncbi.nlm.nih.gov/pubmed/20634905
Giugliani R, Federhen A, Rojas MV, et al. Mucopolysaccharidosis I, II, and VI: brief review and guidelines for treatment. Genet Mol Biol. 2010;33:589-604. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3036139/
Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics. 2007;120:405-418. https://www.ncbi.nlm.nih.gov/pubmed/17671068
INTERNET
Genetics Home Reference. Mucopolysaccharidosis VI. https://ghr.nlm.nih.gov/condition/mucopolysaccharidosis-type-vi Accessed July 15, 2020
Harmatz P, Nicely H, Tuberville S, Valayannopoulos V. Mucopolysaccharidosis type 6. Orphanet Encyclopedia, April 2010. Available at https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=24&Disease_Disease_Search_diseaseGroup=Maroteaux-Lamy-disease&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Mucopolysaccharidosis-type-6&title=Mucopolysaccharidosis%20type%206&search=Disease_Search_Simple Accessed July 15, 2020.
McKusick VA., ed. Online Mendelian Inheritance in Man (OMIM). Baltimore. MD: The Johns Hopkins University; Entry No:253200; Last Update: 01/21/2020. Available at: https://omim.org/entry/253200 Accessed July 15, 2020.

NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportOnline Mendelian Inheritance In Man (OMIM) has a summary of published research about this condition and includes references from the medical literature. The summary contains medical and scientific terms, so we encourage you to share and discuss this information with your doctor. OMIM is authored and edited at the McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine.
View reportMedlinePlus has information about this condition that may include a description, frequency, causes, inheritance, and links to more information. The information is written for the public, including patients, caregivers and families. MedlinePlus is a service of the National Library of Medicine (NLM), which is part of the National Institutes of Health (NIH).
View reportPlease complete this form to access the requested resource.

