

Last updated:
1/23/2024
Years published: 1987, 1988, 1989, 1990, 1998, 2004, 2007, 2017, 2020, 2024
NORD gratefully acknowledges Lisa Melton, PhD, Head of Research, Sanfilippo Children’s Foundation, Australia, for assistance in the preparation of this report.
Summary
Mucopolysaccharidosis type III (MPS III) is a rare genetic condition that causes fatal brain damage. It is also known as Sanfilippo syndrome and is a type of childhood dementia.
MPS III is caused by the lack of an enzyme that normally breaks down and recycles a large, complex sugar molecule called heparan sulfate. This heparan sulphate accumulates and causes damage to the cells of the body, particularly in the central nervous system (brain and spinal cord).
There are four subtypes of MPS III: types A, B, C and D. Each type is caused by a change (variant or mutation) in a different gene (see below). All types of MPS III are associated with mental deterioration, but the severity and rate of progression depends on the type of MPS III. There is also variability in severity within the subtypes and even between affected siblings.
Introduction
Sanfilippo syndrome belongs to a group of disorders known as the mucopolysaccharidoses (MPS), which are part of a larger group of disorders known as lysosomal storage disorders.
Children with MPS III usually appear healthy at birth, but developmental delay is usually evident by age 2-5 years. A mild speech delay is often one of the first presenting features. Mental and motor development peak by 3-6 years of age, after which intellectual decline usually occurs Severe behavioral disturbances, such as hyperactivity and irritability, are a very common feature of Sanfilippo syndrome, and one of the more difficult aspects of the disorder to manage. The nature of the behavioral symptoms can often lead to an initial diagnosis (misdiagnosis) of autism and/or attention deficit/hyperactivity disorder (ADHD).
Other symptoms can include coarse hair, excess hair growth (hirsutism), slightly coarse facial features, severe sleeping problems, mildly enlarged liver and/or spleen, speech delay, respiratory and ear infections, diarrhea, hernias, seizures and a wobbly and erratic walk. The heart may also be affected and children with MPS III also often have hearing loss and vision impairment.
Children with Sanfilippo syndrome usually start to lose their intellectual functions, especially speech, before their motor function declines. Death can occur from before the age of 10 or not until the third or fourth decades of life, with the average being around 15 to 20 years of age. Children with MPS IIIC have a longer life expectancy into the mid-twenties on average. There are other forms of MPS III which result in slower progression and a longer life expectancy.
All four types of MPS III are caused by variants in different genes that contain the instructions for making enzymes that break down heparan sulfate.
TYPE..……….GENE…………ENZYME
MPS IIIA……..SGSH…………heparan N-sulfatase
MPS IIIB……..NAGLU……….alpha-N-acetylglucosaminidase
MPS IIIC……..HGSNAT……..heparan-alpha-glucosaminide N-acetyltransferase
MPS IIID……..GNS…………..N-acetylglucosamine 6-sulfatase
MPS III is inherited in an autosomal recessive manner. This means that both parents have one copy of the altered gene and one normal copy – they are known as carriers and do not show signs of the condition. A child with MPS III inherits two copies of the altered gene, one from each parent.
In autosomal recessive inheritance, in each pregnancy of a couple who are both carriers, there is a:
– 25% (1 in 4) chance of having an affected child
– 50% (1 in 2) chance of a child receiving only one copy of the altered gene and therefore being a carrier
– 25% (1 in 4) chance that a child will be neither affected nor a carrier.
The risk is the same for males and females.
The combined estimated prevalence of Sanfilippo syndrome (types A, B, C and D) is between 1:50,000 and 1:250,000 depending on the population studied. Type A is the most common globally; however, the prevalence of subtypes can vary depending on the region, with type A being more prevalent in Northern Europe and Eastern Europe than in the Mediterranean countries. Type B is the most prevalent subtype in Southern Europe. Types C and D are much less common overall, with estimated global incidences of 1:1,500,000 and 1:1,000,000 respectively.
To diagnose MPS III, mucopolysaccharides are usually first measured in urine, followed by measurement of enzyme activity in blood or a small skin sample. Increased heparan sulfate in urine, and a decrease in the activity of any one of the four enzymes (shown in the table above) is usually consistent with a diagnosis of MPS III and will identify the MPS III type (A, B, C or D). It is important to know the MPS III type as many of the treatments being developed are only for specific types.
Genetic testing of a blood sample will allow the identification of the exact changes in the DNA. It is important to attend genetic counseling to learn the implications for other children in the family, future pregnancies and extended family members. The counselor will explain the inheritance pattern and help advise who should be tested.
If the genetic diagnosis is known, this information can be used to test other at-risk members of the family. It can also be used for prenatal testing of future pregnancies (testing a fetus while still in the womb) and/or preimplantation diagnosis (testing of embryos created through IVF to select those that do not carry the relevant gene mutation).
Treatment
Treatment of Sanfilippo syndrome is symptomatic and supportive. It is important for children with MPS III to be managed by a multidisciplinary team of specialists to give these children the best quality of life. At different stages this could include a combination of the following: a neurologist, developmental pediatrician, metabolic/genetics specialist, orthopedist, gastroenterologist, ophthalmologist, cardiologist, endocrinologist, allied health (e.g. physiotherapy, OT, behavioral therapists, speech therapist) and an ENT (ear, nose and throat) specialist.
The Consensus Guidelines for Sanfilippo Syndrome Clinical Care were published in 2022 and provide comprehensive evidence-based, expert-led recommendations for healthcare providers and families on how to approach Sanfilippo syndrome-specific management and monitoring of disease-related changes.
Clinical trials designed to gauge the safety and efficacy of several different approaches are under way. Therapeutic approaches in clinical trial include:
– Gene therapy which involves using a harmless virus to deliver a functional copy of the altered gene into the body
– Gene modified stem cell therapy, where stem cells are removed from the body, the genetic error corrected and the stem cells transplanted back into the body
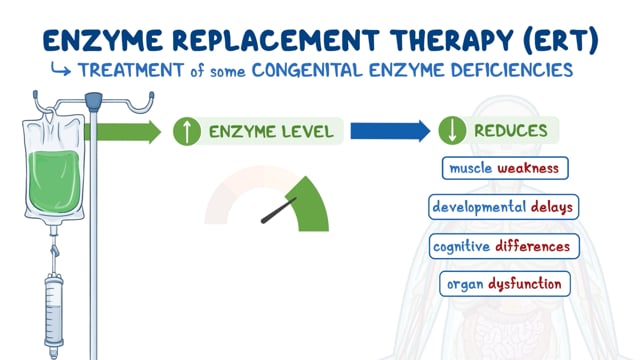
– Enzyme replacement therapy, where the missing enzyme is administered
Laboratory based research is also searching for drugs to help slow disease progression and improve quality of life. It is thought that a combination of treatments will be required to give the best outcome.
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
For information about clinical trials being conducted at the NIH Clinical Center in Bethesda, MD, contact the NIH Patient Recruitment Office:
Toll-free: (800) 411-1222
TTY: (866) 411-1010
Email: [email protected]
Some current clinical trials also are posted on the following page on the NORD website: https://rarediseases.org/living-with-a-rare-disease/find-clinical-trials/
For information about clinical trials sponsored by private sources, contact: www.centerwatch.com
For more information about clinical trials conducted in Europe, contact: https://www.clinicaltrialsregister.eu/
Sanfilippo Children’s Foundation (Australia) and Cure Sanfilippo Foundation (USA) maintain up to date information on Sanfilippo syndrome clinical trials on their websites here:
https://www.sanfilippo.org.au/research/therapy-avenues
TEXTBOOKS
Fuller M, Meikle PJ, Hopwood JJ. Epidemiology of lysosomal storage diseases: an overview. In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006;Chapter 2.
Clarke JTR. Mucopolysaccharide Storage (MPS) Diseases. In: NORD Guide to Rare Disorders. Lippincott Williams & Wilkins. Philadelphia, PA. 2003:474-79.
Neufeld EF, Muenzer J. The Mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, et al. Eds. The Metabolic Molecular Basis of Inherited Disease. 8th ed. McGraw-Hill Companies. New York, NY; 2001:3421-41.
JOURNAL ARTICLES
Muschol N, Giugliani R, Jones SA, et al. Sanfilippo syndrome: consensus guidelines for clinical care. Orphanet J Rare Dis. 2022;17(1):391. Published 2022 Oct 27. doi:10.1186/s13023-022-02484-6
Pearse Y, Iacovino M. A cure for Sanfilippo syndrome? a summary of current therapeutic approaches and their promise. Med Res Arch. 2020;8(2):10.18103/mra.v8i2.2045. doi:10.18103/mra.v8i2.2045
Zelei T, Csetneki K, Vokó Z, Siffel C. Epidemiology of Sanfilippo syndrome: results of a systematic literature review. Orphanet J Rare Dis. 2018;13(1):53. Published 2018 Apr 10. doi:10.1186/s13023-018-0796-4
Andrade F, Aldámiz-Echevarría L, Llarena M, Couce ML. Sanfilippo syndrome: Overall review. Pediatr Int. 2015 Jun;57(3):331-8.
Fedele AO. Sanfilippo syndrome: causes, consequences, and treatments. Appl Clin Genet. 2015;8:269-81.
Valstar MJ, Ruijter GJ, van Diggelen OP, Poorthuis BJ, Wijburg FA. Sanfilippo syndrome: a mini-review. J Inherit Metab Dis. 2008;31(2):240–52.
INTERNET
Mucopolysaccharidosis Type IIIA. Online Mendelian Inheritance in Man (OMIM). The Johns Hopkins University. Entry Number; 252900: Last Edit Date: 2/17/22.Available at https://www.omim.org/entry/252900?search=252900&highlight=252900 Accessed January 18, 2024.
Mucopolysaccharidosis Type IIIB. Online Mendelian Inheritance in Man (OMIM). The Johns Hopkins University. Entry Number; 252920: Last Edit Date: 10/10/2023. Available at: https://www.omim.org/entry/252920?search=252920&highlight=252920 Accessed January 18, 2024.
Mucopolysaccharidosis Type IIIC. Online Mendelian Inheritance in Man (OMIM). The Johns Hopkins University. Entry Number; 252930: Last Edit Date: 06/13/2022. Available at: https://www.omim.org/entry/252930?search=252930&highlight=252930 Accessed January 18, 2024.
Mucopolysaccharidosis Type IIID. Online Mendelian Inheritance in Man (OMIM). The Johns Hopkins University. Entry Number; 252940: Last Edit Date; 02/17/2022. Available at: https://www.omim.org/entry/252940?search=252940&highlight=252940 Accessed January 18, 2024.
Defendi GL. Genetics of Mucopolysaccharidosis Type III. Medscape. Updated: April 19, 2023. www.emedicine.com/ped/topic2040.htm Accessed January 18, 2024.
A Guide to Understanding Sanfilippo syndrome (mucopolysaccharidosis type III; MPS III). MPS Australia. https://www.mpssociety.org.au/about-the-mps-and-related-diseases/mps-iii/ Accessed January 18, 2024.
MPS III (Sanfilippo syndrome) National MPS Society. https://mpssociety.org/learn-about-mps/diseases/mps-iii/ Accessed January 18, 2024.

NORD strives to open new assistance programs as funding allows. If we don’t have a program for you now, please continue to check back with us.
NORD and MedicAlert Foundation have teamed up on a new program to provide protection to rare disease patients in emergency situations.
Learn more https://rarediseases.org/patient-assistance-programs/medicalert-assistance-program/Ensuring that patients and caregivers are armed with the tools they need to live their best lives while managing their rare condition is a vital part of NORD’s mission.
Learn more https://rarediseases.org/patient-assistance-programs/rare-disease-educational-support/This first-of-its-kind assistance program is designed for caregivers of a child or adult diagnosed with a rare disorder.
Learn more https://rarediseases.org/patient-assistance-programs/caregiver-respite/The information provided on this page is for informational purposes only. The National Organization for Rare Disorders (NORD) does not endorse the information presented. The content has been gathered in partnership with the MONDO Disease Ontology. Please consult with a healthcare professional for medical advice and treatment.
The Genetic and Rare Diseases Information Center (GARD) has information and resources for patients, caregivers, and families that may be helpful before and after diagnosis of this condition. GARD is a program of the National Center for Advancing Translational Sciences (NCATS), part of the National Institutes of Health (NIH).
View reportOrphanet has a summary about this condition that may include information on the diagnosis, care, and treatment as well as other resources. Some of the information and resources are available in languages other than English. The summary may include medical terms, so we encourage you to share and discuss this information with your doctor. Orphanet is the French National Institute for Health and Medical Research and the Health Programme of the European Union.
View reportPlease complete this form to access the requested resource.

